დოკუმენტის სტრუქტურა
განმარტებების დათვალიერება
დაკავშირებული დოკუმენტები
დოკუმენტის მონიშვნები
 უკან დაბრუნება
უკან დაბრუნება | ცხოველის საკვების სახელმწიფო კონტროლისათვის ნიმუშის აღებისა და გამოკვლევის მეთოდების დამტკიცების შესახებ | |
|---|---|
| დოკუმენტის ნომერი | 107 |
| დოკუმენტის მიმღები | საქართველოს მთავრობა |
| მიღების თარიღი | 02/03/2022 |
| დოკუმენტის ტიპი | საქართველოს მთავრობის დადგენილება |
| გამოქვეყნების წყარო, თარიღი | ვებგვერდი, 14/03/2022 |
| ძალაში შესვლის თარიღი | 01/01/2024 |
| სარეგისტრაციო კოდი | 240110000.10.003.023317 |
|
ცხოველის საკვების სახელმწიფო კონტროლისათვის ნიმუშის აღებისა და გამოკვლევის მეთოდების დამტკიცების შესახებ
|
| მუხლი 1 |
სურსათის/ცხოველის საკვების უვნებლობის, ვეტერინარიისა და მცენარეთა დაცვის კოდექსის 75-ე მუხლის მე-2 ნაწილის შესაბამისად, დამტკიცდეს თანდართული „ცხოველის საკვების სახელმწიფო კონტროლისათვის ნიმუშის აღებისა და გამოკვლევის მეთოდები“.
|
| მუხლი 2 |
დადგენილება ამოქმედდეს 2024 წლის 1 იანვრიდან.
|
|
ცხოველის საკვების სახელმწიფო კონტროლისათვის ნიმუშის აღებისა და გამოკვლევის მეთოდები
|
მუხლი 1
1. ცხოველის საკვების სახელმწიფო კონტროლისთვის, კერძოდ, შემადგენლობის განსაზღვრა (მათ შორის, მასალის, რომელიც შეიცავს ან შედგება ან წარმოებულია გენმოდიფიცირებული ორგანიზმებისაგან (გმო), „ცხოველთა კვებაში გამოსაყენებელი ცხოველის საკვები დანამატების წესის დამტკიცების შესახებ“ საქართველოს მთავრობის დადგენილებით განსაზღვრული ცხოველის საკვები დანამატების და საქართველოს კანონმდებლობით განსაზღვრული არასასურველი ნივთიერებების) ხორციელდება „ცხოველის საკვების სახელმწიფო კონტროლისათვის ნიმუშის აღებისა და გამოკვლევის მეთოდების“ (შემდგომში – წესი) დანართით №1 „ნიმუშის აღების მეთოდები“ განსაზღვრული მეთოდების შესაბამისად. 2. ცხოველის საკვებში საქართველოს კანონმდებლობით განსაზღვრული პესტიციდების ნაშთი ოდენობის შესაბამისობის კონტროლისთვის გამოიყენება ამ წესის დანართით №1 განსაზღვრული ნიმუშის აღების მეთოდი. მუხლი 2 ნიმუშების მომზადება გამოკვლევისათვის და შედეგების გამოხატვა ხორციელდება ამ წესის დანართით №2 – „ზოგადი დებულებები ცხოველის საკვების გამოკვლევის მეთოდების შესახებ“ განსაზღვრული მეთოდების შესაბამისად. მუხლი 3 ცხოველის საკვების სახელმწიფო კონტროლი უნდა განხორციელდეს ამ წესის დანართით №3 – „ცხოველის საკვების მასალისა და კომბინირებული საკვების შემადგენლობის კონტროლისათვის გამოყენებული გამოკვლევის მეთოდები“, დანართით №4 – „ცხოველის საკვებში ავტორიზებული დანამატების შემცველობის კონტროლისათვის გამოყენებული გამოკვლევის მეთოდები“, დანართით №5 – „ცხოველის საკვებში არასასურველი ნივთიერებების კონტროლის გამოკვლევის მეთოდები“ და დანართით №6 – „ცხოველის საკვების კონტროლისას ცხოველური წარმოშობის შემადგენელი ნაწილაკების განსაზღვრასთან დაკავშირებული გამოკვლევის მეთოდები“ განსაზღვრული მეთოდებით. მუხლი 4 ფრინველის კომბინირებული საკვების ენერგეტიკული ღირებულება გამოთვლილი უნდა იქნეს ამ წესის დანართის №7 „ფრინველის საკვების ენერგეტიკული ღირებულების გამოანგარიშების მეთოდი“ შესაბამისად. მუხლი 5 უკვე ავტორიზაციის არმქონე ცხოველის საკვების დანამატის არალეგალური (უკანონო) შემცველობის კონტროლისათვის გამოკვლევის მეთოდები განსაზღვრულია ამ წესის დანართით №8 „გამოკვლევის მეთოდები ცხოველის საკვებში უკვე ავტორიზაციის არმქონე, უკანონოდ არსებული, ცხოველის საკვები დანამატების კონტროლისთვის“.
დანართი № 1 ნიმუშის აღების მეთოდები 1. მიზანი და რეგულირების სფერო 1.1. ცხოველის საკვების სახელმწიფო კონტროლისათვის განკუთვნილი ნიმუშების აღება უნდა განხორციელდეს ამ დანართით განსაზღვრული მოთხოვნების შესაბამისად. ამ მოთხოვნების შესაბამისად აღებული ნიმუშები მიიჩნევა, როგორც შერჩეული პორციის რეპრეზენტატიულ ნიმუშებად. 1.2. რეპრეზენტატული ნიმუშის აღების მიზანია ლოტიდან მცირე ფრაქციის მოპოვება ისე, რომ ამ ფრაქციის რომელიმე კონკრეტული მახასიათებლის განსაზღვრა წარმოადგენდეს ლოტის მახასიათებლის საშუალო მაჩვენებელს. ლოტიდან ნიმუშის აღება ხდება ლოტის სხვადასხვა ცალკეული პოზიციებიდან ინკრემენტალური ნიმუშების მრვალაჯერადი აღებით. ეს ინკრემენტალური ნიმუშები უნდა გაერთიანდეს შერევით რათა მოხდეს გაერთიანებული ნიმუშის ფორმირება, საიდანაც მომზადდება რეპრეზენტატული საბოლოო ნიმუშები რეპრეზენტატული გაყოფით. 1.3. თუ ვიზუალური შემოწმებით, ცხოველის საკვების ის პორცია, საიდანაც უნდა მოხდეს ნიმუშის აღება, ჩანს, რომ ხარისხობრივად განსხვავება იმავე ლოტის დანარჩენი ცხოველის საკვებისაგან, უნდა მოხდეს ასეთი პორციების გამოიყოფა დანარჩენი ცხოველის საკვებისგან და უნდა ჩაითვალოს, როგორც ცალკე არსებული ქველოტად. თუ შეუძლებელია ცხოველის საკვების დაყოფა ცალკე ქველოტად, ცხოველის საკვებიდან ნიმუში იღება, როგორც ერთი ლოტიდან. ასეთ შემთხვევებში, ფაქტი უნდა აღინიშნოს ნიმუშის აღების ოქმში. 1.4. თუ დადგინდება, რომ ცხოველის საკვები, რომლიდანაც ნიმუშის აღება განხორციელდა ამ წესით განსაზღვრული მოთხოვნების შესაბამისად, ვერ აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს, მიეკუთვნება იმავე ცხოველის საკვების ლოტის ნაწილის კლასს ან აღწერილობას, უნდა ჩაითვალოს, რომ ამ ლოტს მიკუთვნებული მთლიანი ცხოველის საკვები ისეა დაზიანებული, რომ ლოტის დარჩენილი ნაწილიც ვერ აკმაყოფილებს საქართველოს კანონმდებლობაით განსაზღვრულ მოთხოვნებს, გარდა იმ შემთხვევებისა, თუკი დეტალური შეფასების შემდეგ ვერ იქნება მოპოვებული ასეთი მტკიცებულება. 2. ტერმინთა განმარტებები ამ წესის მიზნებისათვის გამოყენებულ ტერმინებს აქვს შემდეგი მნიშვნელობები: 2.1. ლოტი (ან პარტია) – ცხოველის საკვების იდენტიფიცირებული რაოდენობა, რომელსაც გააჩნია საერთო მახასიათებლები, ისეთი როგორიცაა, წარმოშობა, ნაირსახეობა, შეფუთვის ტიპი, შემფუთავი, ტვირთის გამგზავნი ან ეტიკეტირება, ხოლო, წარმოების პროცესის შემთხვევაში, წარმოებული ერთეული ერთი საამქროდან, რომელიც იყენებს ერთგვაროვანი წარმოების პარამეტრებს ან რამდენიმე ასეთი ერთეული, რომელიც წარმოებულია უწყვეტად და შენახულია ერთად; 2.2. შერჩეული პორცია – ლოტი, ლოტის იდენტიფიცირებული ნაწილი ან ქველოტი; 2.3. დალუქული ნიმუში – ნიმუში დალუქულია ისე, რომ შეუძლებელია ნიმუშზე ყოველგვარი წვდომა ლუქის მოხსნის ან დაზიანების/გატეხვის გარეშე; 2.4. ინკრემენტალური ნიმუში – შერჩეული პორციის ერთი წერტილიდან აღებული რაოდენობა; 2.5. გაერთიანებული ნიმუში – იმავე შერჩეული პორციიდან აღებული ინკრემენტალური ნიმუშების ერთობლიობა; 2.6. შემცირებული ნიმუში – გაერთიანებული ნიმუშის ნაწილი, რომელიც ამ უკანასკნელისგან მიიღება რეპრეზენტატული შემცირებით; 2.7. საბოლოო ნიმუში – შემცირებული ნიმუშის ან ჰომოგენიზებული გაერთიანებული ნიმუშის ნაწილი; 2.8. ლაბორატორიული ნიმუში – ლაბორატორიისათვის განკუთვნილი ნიმუში (როგორც მიიღო ლაბორატორიამ) და შეიძლება იყოს საბოლოო, შემცირებული ან გაერთიანებული ნიმუში. 3. ზოგადი დებულებები 3.1. ნიმუშის აღებას ახორციელებს სსიპ – სურსათის ეროვნული სააგენტოს (შემდგომში – სააგენტო) და სსიპ – შემოსავლების სამსახურის მიერ ამ მიზნით უფლებამოსილი პირები. 3.2. ნიმუში უნდა იყოს დალუქული ისე, რომ თავიდან იქნეს აცილებული ნიმუშზე წვდომა ლუქის დაზიანების/გატეხვის ან მოხსნის გარეშე. ლუქის აღნიშვნა უნდა იყოს მკაფიოდ იდენტიფიცირებადი და ადვილად ხილვადი. ალტერნატივის სახით, ნიმუში შეიძლება მოთავსდეს ისეთ ჭურჭელში, რომელიც იხურება ისე, რომ მისი გახსნა შეუძლებელია ჭურჭლის ან კონტეინერის შეუქცევადად დაზიანების გარეშე, ჭურჭლის ან კონტეინერის ხელახალი გამოყენების თავიდან ასაცილებლად. 3.3. ნიმუშის იდენტიფიკაციისთვის, ნიმუში უნდა იყოს მარკირებული ისე, რომ არ მოხდეს მისი წაშლა და იდენტიფიკაცია უნდა მოხდეს ისე, რომ ერთმნიშვნელოვანი კავშირი იყოს ნიმუშის აღების ოქმთან. 3.4. თითოეული გაერთიანებული ნიმუშიდან ხდება სულ მცირე ორი საბოლოო ნიმუშის აღება: არანაკლებ ერთი – კონტროლისთვის (აღსრულებისთვის) და ერთი – ცხოველის საკვების ბიზნესოპერატორისთვის (დაცვისთვის). ასევე, ერთი საბოლოო ნიმუში შეიძლება აღებულ იქნეს რეფერენს ნიმუშად. სრული გაერთიანებული ნიმუშის ჰომოგენიზაციის შემთხვევაში, საბოლოო ნიმუშები მიიღება ჰომოგენიზებული გაერთიანებული ნიმუშიდან. 4. ხელსაწყოები/აღჭურვილობა 4.1. ნიმუშის ასაღები ხელსაწყო დამზადებული უნდა იყოს ისეთი მასალებისგან, რომელიც ვერ დააბინძურებს გამოსაკვლევ პროდუქტს. მრავალჯერადი მოხმარებისთვის განკუთვნილი ხელსაწყო ადვილად უნდა იწმინდებოდეს ჯვარედინი დაბინძურების თავიდან აცილების მიზნით. 4.2. ხელსაწყო, რომელიც რეკომენდებულია ცხოველის მყარი საკვების ნიმუშის ასაღებად: 4.2.1. ნიმუშის ხელით აღების შემთხვევაში: 4.2.1.1. ბრტყელძირიანი ნიჩაბი ვერტიკალური გვერდებით; 4.2.1.2. ნიმუშის ასაღები შუბი გრძელი ხვრელებითა ან სექციებით. ნიმუშის ასაღები შუბის ზომები უნდა შეესაბამებოდეს შერჩეული პორციის მახასიათებლებს (კონტეინერის სიღრმე, ტომარის ზომები და ა.შ.) და ცხოველის საკვების ნაწილაკის (ერთეულის) ზომას. იმ შემთხვევაში, თუ ნიმუშის ასაღებ შუბს აქვს მრავალი ხვრელი, რათა უზრუნველყოს ნიმუშის აღება ზონდის გასწვრივ სხვადასხვა ადგილას, ხვრელები უნდა გამოიყოს სექციებით ან მრავალსაფეხურიანი ხვრელებით. 4.2.2. ნიმუშის მექანიკურად აღების შემთხვევაში: 4.2.2.1 შესაბამისი მექანიკური ხელსაწყო შეიძლება გამოყენებულ იქნეს გადმოსატვირთი ცხოველის საკვების ნიმუშის ასაღებად. შესაბამისი ნიშნავს, რომ სულ მცირე, გადმოტვირთვის მთლიან ნაკადზე ჩატარდა ნიმუშის აღება; 4.2.2.2 გადმოტვირთვაში მყოფი ცხოველის საკვებიდან ნიმუშის აღება (გადმოტვირთვის მაღალ სიჩქარეზე) შეიძლება შესრულდეს ავტომატური ნიმუშის ამღები ხელსაწყოებით. 4.2.3. გამყოფი თუ ეს შესაძლებელია და მისაღებია, რეპრეზენტატული წესების დაცვით შემცირებული ნიმუშების მოსამზადებლად გამოყენებული უნდა იქნეს ის მოწყობილობა, რომელიც შექმნილია ნიმუშების, შეძლებისდაგვარად თანაბარ ნაწილებად, დასაყოფად. 5. მოთხოვნები ინკრემენტალური ნიმუშის რაოდენობასთან დაკავშირებით
5.1. ცხოველის საკვებში თანაბრად გადანაწილებული ნივთიერებების ან პროდუქტების კონტროლისთვის ინკრემენტალური ნიმუშთან დაკავშირებული რაოდენობრივი მოთხოვნები 5.1.1. ცხოველის შეუფუთავი მყარი საკვების შემთხვევაში:
5.1.2. ცხოველის შეუფუთავი თხევადი საკვების შემთხვევაში:
5.1.3. ცხოველის შეფუთული საკვების შემთხვევაში – ცხოველის (მყარი და თხევადი) საკვები შეიძლება შეფუთული იქნეს ჩანთებით, ტომრებით, ბიდონებით, ბარელებით და ა.შ. რომლებიც ცხრილში მითითებულია, როგორც ერთეული. მსხვილი ერთეულების (≥ 500 კგ ან ლიტრი) ნიმუში აღებული უნდა იქნეს იმ მოთხოვნების შესაბამისად, რომლებიც გათვალისწინებულია შეუფუთავი ცხოველის საკვებისთვის (იხ. ამ დანართის 5.1.1 და 5.1.2 პუნქტები).
5.1.4. ცხოველის საკვები ბრიკეტებისა და სალოკის შემთხვევაში სულ მცირე ერთი ბრიკეტი ან სალოკი უნდა იქნეს აღებული 25 შერჩეული პორციიდან, მაგრამ არაუმეტეს 4 ბრიკეტისა ან სალოკისა. საკვები ბრიკეტების ან სალოკისთვის, რომლის წონაც არაუმეტეს 1 კგ-ია, თითოეულის შემთხვევაში, ინკრემენტალური ნიმუში უნდა იყოს ერთი ბრიკეტის ან სალოკის შემცველობა. 5.1.5. უხეში საკვების/ფურაჟის შემთხვევაში:
5.2. ცხოველის საკვებში არათანაბრად გადანაწილებული ნივთიერებების ან კომპონენტების კონტროლისთვის ინკრემენტალური ნიმუშთან დაკავშირებული რაოდენობრივი მოთხოვნები ქვემოთ მითითებული რაოდენობრივი მოთხოვნები ინკრემენტალური ნიმუშების მიმართ გამოიყენება შემდეგ სიტუაციებში:
თუ სააგენტოს აქვს საფუძვლიანი ეჭვი, რომ ასეთი არათანაბარი გადანაწილება ხდება აგრეთვე კომბინირებული საკვებში კომპონენტით ან ნივთიერებით გამოწვეული ჯვარედინი დაბინძურების შემთხვევაშიც, შეიძლება გამოყენებულ იქნეს ქვემოთ მოცემულ ცხრილში მოცემული რაოდენობრივი მოთხოვნები:
5.3. ძალიან დიდი ლოტის შემთხვევებში ინკრემენტალური ნიმუშებთან დაკავშირებული რაოდენობრივი მოთხოვნები დიდი შერჩეული პორციის (შერჩეული პორცია > 500 ტონა) შემთხვევაში, ასაღები ინკრემენტალური ნიმუშების რაოდენობა = 40 ინკრემენტალურ ნიმუშს + √ტონა ნივთიერებების ან პროდუქტების კონტროლთან დაკავშირებით, რომლებიც ერთნაირად ნაწილდება ცხოველის საკვებში ან 100 ინკრემენტალურ ნიმუშს + √ტონა კომპონენტების ან ნივთიერებების კონტროლთან დაკავშირებით, რომლებიც სავარაუდოდ არათანაბრად განაწილდება ცხოველის საკვების მასალებში. 6. რაოდენობრივი მოთხოვნები გაერთიანებულ ნიმუშთან დაკავშირებით 1. გაერთიანებულ ნიმუშთან დაკავშირებით:
(*) თუ ცხოველის საკვების აღებული ნიმუშის ღირებულება მაღალია, მცირე რაოდენობის გაერთიენებული ნიმუში შეიძლება იქნეს აღებული იმ პირობით, თუ ეს აღწერილი და დაფიქსირებული იქნება ნიმუშის აღების ოქმში. (**) გენმოდიფიცირებული მასალის არსებობის კონტროლისთვის, რომლისთვისაც ავტორიზაციის პროცედურა გადაწყვეტილების მოლოდინშია ან რომლის ავტორიზაციის ვადა ამოიწურა, გაერთიანებული ნიმუში უნდა შეიცავდეს სულ მცირე 35000 თესლს/მარცვალს. ეს ნიშნავს, რომ სიმინდისთვის გაერთიანებული ნიმუშის ზომა უნდა იყოს არანაკლებ 10,5 კგ, ხოლო სოიოსთვის 7 კგ. სხვა თესლისა და მარცვლეულისთვის, როგორიცაა ქერი, ფეტვი, შვრია, ბრინჯი, ჭვავის, ხორბლისა და რაფსი, გაერთიანებული ნიმუშის ზომა – 4 კგ. შეესაბამება 35 000-ზე მეტ თესლს. (***) ცხოველის შეფუთული საკვების შემთხვევაში, ასევე შეიძლება ვერ მოხერხდეს 4 კგ ზომის მიღწევა გაერთიანებული ნიმუშისთვის ინდივიდუალური ერთეულების სიდიდიდან გამომდინარე. (****) დაბალი კუთრი წონა უხეში საკვების ან ფურაჟის შემთხვევაში, (მაგ., თივა, ჩალა), გაერთიანებული ნიმუშის მინიმალური წონა უნდა იყოს 1 კგ. 6. რაოდენობრივი მოთხოვნები საბოლოო ნიმუშთან დაკავშირებით არანაკლებ ერთი საბოლოო ნიმუშის გამოკვლევაა საჭირო. საბოლოო ნიმუშიდან გამოსაკვლევი რაოდენობა არ შეიძლება იყოს ქვემოთ მოცემულ რაოდენობაზე ნაკლები:
(*) გენმოდიფიცირებული მასალის არსებობის გასაკონტროლებლად, რომლისთვისაც ავტორიზაციის პროცედურა გადაწყვეტილების მოლოდინშია ან რომლის ავტორიზაციის ვადა ამოიწურა, საბოლოო ნიმუში უნდა შეიცავდეს არანაკლებ 10,000 თესლს/მარცვალს. ეს ნიშნავს, რომ სიმინდისთვის, საბოლოო ნიმუშის წონა უნდა იყოს სულ მცირე 3000 გ, ხოლო სოიოსთვის – 2000 გ. სხვა თესლისა და მარცვლეულისთვის, როგორიცაა ქერი, ფეტვი, შვრია, ბრინჯი, ჭვავი, ხორბალი და რაფსი, 500-გრამიანი საბოლოო ნიმუში 10,000-ზე მეტ თესლს შეესაბამება. (**) იმ შემთხვევაში, თუ გაერთიანებული ნიმუშის სიდიდე მნიშვნელოვნად ნაკლებია 4 კგ-ზე ან ლიტრზე (იხ. ამ დანართის მე-6 პუნქტის სქოლიოები), ასევე შეიძლება აღებულ იქნეს საბოლოო ნიმუშის უფრო მცირე რაოდენობა იმ პირობით, თუ ეს აღწერილი და დაფიქსირებული იქნება ნიმუშის აღების ოქმში. (***) პესტიციდების ნაშთი ოდენობის (ნარჩენების) დადგენის მიზნით, პარკოსანი კულტურების, მარცვლეულის მარცვლებისა და კაკლოვანი კულტურების ნიმუშის აღების შემთხვევაში, საბოლოო ნიმუშის მინიმალური წონა უნდა იყოს 1 კგ. 8. ნიმუშის აღების მეთოდი ძალიან დიდი ლოტისთვის ან დასაწყობებული ან სატრანსპორო საშუალებაში მყოფი ლოტისთვის, როდესაც მთლიანი ლოტიდან ნიმუშის აღება შეუძლებელია 8.1 ზოგადი პრინციპები თუ სატრანსპორტო საშუალებაში ან საწყობში ტვირთი ისეა განთავსებული, რომ შეუძლებელია მთლიანი ლოტიდან (სხვადასხვა ადგილებიდან) ინკრემენტალური ნიმუშების აღება, ნიმუშების აღება უნდა მოხდეს გადმოტვირთვისას. დიდი საწყობის შემთხვევაში, რომლებიც განკუთვნილია ცხოველის საკვების შესანახად, ოპერატორები წახალისებული უნდა იყვნენ აღჭურვონ საწყობები ნიმუშის ავტომატური აღების მოწყობილობით, რითაც შესაძლებელი იქნება მოხდეს შენახული ტვირთის მთლიანი ლოტიდან (სხვადასხვა ადგილებიდან) ნიმუშის აღება (ავტომატურად). ამ დანართის ამ პუნქტში (მე-8 პუნქტი) გათვალისწინებული ნიმუშების აღების პროცედურების გამოყენების შემთხვევაში, ცხოველის საკვების ბიზნესოპერატორი ან მისი წარმომადგენელი ინფორმირებული უნდა იყოს ნიმუშის აღების პროცედურის შესახებ. იმ შემთხვევაში, თუ ცხოველის საკვების ბიზნესოპერატორი ან მისი წარმომადგენელი ნიმუშის აღების პროცედურას ეჭვქვეშ აყენებს, ცხოველის საკვების ბიზნესოპერატორი ან მისი წარმომადგენელის ხარჯით სააგენტო იღებს ნიმუშებს ტვირთის მთლიანი ლოტის სხვადასხვა ადგილებიდან. 8.2. გემით ტრანსპორტირებული დიდი ლოტები 8.2.1. გემით ტრანსპორტირებული დიდი ლოტიდან ნიმუშის დინამიური აღება გემებში დიდი ლოტებიდან ნიმუშის აღება სასურველია ხორციელდებოდეს პროდუქტის გადმოტვირთვის დროს (ნიმუშის დინამიური აღება). ნიმუშის აღება უნდა ჩატარდეს თითო სათავსოსთან/კონტეინერთან მიმართებაში (კონტეინერი, რომლის ფიზიკური განცალკევებაა შესაძლებელი). თუმცა, სათავსოები/კონტეინერები ნაწილობრივ ცარიელდება ერთმანეთის მიყოლებით ისე, რომ თავდაპირველ ფიზიკურ განცალკევებას აღარ აქვს ადგილი შემნახველ სათავსოში (ობიექტში) გადატანის შემდეგ. აქედან გამომდინარე, ნიმუშის აღება შეიძლება განხორციელდეს თავდაპირველი ფიზიკური განცალკევებისას ან შემნახველ სათავსოში (ობიექტებში) გადატანის შემდეგ განცალკევებისას. გემის გადმოტვირთვა შეიძლება რამდენიმე დღე გაგრძელდეს. როგორც წესი, ნიმუშის აღება უნდა განხორციელდეს რეგულარული ინტერვალებით, გადმოტვირთვის მთელი პერიოდის განმავლობაში. თუმცა, ყოველთვის არ არის შესაძლებელი ან მიზანშეწონილი სახელმწიფო ინსპექტორის დასწრება ნიმუშების აღების გადმოტვირთვის მთლიან პროცესში. ამრიგად, ნიმუშების აღება დასაშვებია მთლიანი ლოტის ნაწილისგან (შერჩეული პორციიდან). ინკრემენტალური ნიმუშების რაოდენობა განისაზღვრება შერჩეული პორციის ზომის (სიდიდის) გათვალისწინებით. ერთი და იმავე კლასის და აღწერილობის ცხოველის საკვების ლოტის ნაწილიდან ნიმუშის აღებისას, თუ გამოვლინდება, რომ ლოტის ეს ნაწილი არ აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს, მაშინ ჩაითვლება რომ ამ ლოტის ცხოველის მთლიანი საკვები ისეთივეა და ლოტის დარჩენილი ნაწილიც არ აკმაყოფილებს აღნიშნულ მოთხოვნებს, გარდა იმ შემთხვევისა, როდესაც დეტალური შეფასების შემდეგ იქნება მოპოვებული მტკიცებულება იმის შესახებ, რომ დარჩენილი ნაწილი აკმაყოფილებს კანონმდებლობით განსაზღვრულ მოთხოვნებს. მაშინაც კი, თუ სახელმწიფო ნიმუშის აღება ხდება ავტომატურად, ინსპექტორის ყოფნა აუცილებელია. იმ შემთხვევაში, თუ ავტომატური აღება ხდება წინასწარ განსაზღვრული პარამეტრებით, რომელთა შეცვლა შეუძლებელია ნიმუშის აღების დროს და ინკრემენტალური ნიმუშების შეგროვება ხდება დალუქულ ჭურჭელში/ტარაში, რაც ხელს უშლის შესაძლო თაღლითობას, მაშინ ინსპექტორის ყოფნა საჭიროა მხოლოდ ნიმუშის აღების დასაწყისში, ყოველ ჯერზე, როდესაც საჭიროა ნიმუშის ჭურჭლის შეცვლა და ნიმუშის აღების დასრულებისას. 8.2.2. გემით ტრანსპორტირებული ლოტებიდან ნიმუშის აღება სტატიკური გზით იმ შემთხვევაში, თუ ნიმუშის აღება ხორციელდება სტატიკური გზით, გამოყენებული უნდა იყოს იმავე პროცედურა, რაც გათვალისწინებულია შემნახველი სათავსოებისთვის/ობიექტებისთვის (სასილოსეებისთვის), ზევიდან წვდომის გათვალისწინებით (იხ. ამ დანართის პუნქტი 8.4.1). ნიმუშის აღება უნდა მოხდეს ლოტის/სათავსოს ხელმისაწვდომი ნაწილიდან (ზევიდან). ინკრემენტალური ნიმუშების რაოდენობა განისაზღვრება შერჩეული პორციის ზომის (სიდიდის) გათვალისწინებით. ერთი და იმავე კლასის და აღწერილობის ცხოველის საკვების ლოტის ნაწილიდან ნიმუშის აღებისას, თუ გამოვლინდება, რომ ლოტის ეს ნაწილი არ აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს, მაშინ ჩაითვლება რომ ამ ლოტის ცხოველის მთლიანი საკვები ისეთივეა და ლოტის დარჩენილი ნაწილიც არ აკმაყოფილებს აღნიშნულ მოთხოვნებს, გარდა იმ შემთხვევისა, როდესაც დეტალური შეფასების შემდეგ იქნება მოპოვებული მტკიცებულება იმის შესახებ, რომ დარჩენილი ნაწილი აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს. 8.3. საწყობებში შენახული დიდი ლოტებიდან ნიმუშის აღება ნიმუშის აღება უნდა შესრულდეს ლოტის ხელმისაწვდომი ნაწილიდან. ინკრემენტალური ნიმუშების რაოდენობა განისაზღვრება შერჩეული პორციის ზომის (სიდიდის) გათვალისწინებით. ერთი და იმავე კლასის და აღწერილობის ცხოველის საკვების ლოტის ნაწილიდან ნიმუშის აღებისას, თუ გამოვლინდება, რომ ლოტის ეს ნაწილი არ აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს, მაშინ ჩაითვლება რომ ამ ლოტის ცხოველის მთლიანი საკვები ისეთივეა და ლოტის დარჩენილი ნაწილიც არ აკმაყოფილებს აღნიშნულ მოთხოვნებს, გარდა იმ შემთხვევისა, როდესაც დეტალური შეფასების შემდეგ იქნება მოპოვებული მტკიცებულება იმის შესახებ, რომ დარჩენილი ნაწილი აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს. 8.4. ნიმუშის აღება შემნახველ სათავსოში (სასილოსე) 8.4.1. სასილოსეში ნიმუშის აღება, რომელთანაც წვდომა შესაძლებელია (მარტივად) ზემოდან ნიმუშის აღება უნდა შესრულდეს ლოტის ხელმისაწვდომი ნაწილიდან. ინკრემენტალური ნიმუშების რაოდენობა განისაზღვრება შერჩეული პორციის ზომის (სიდიდის) გათვალისწინებით. ერთი და იმავე კლასის და აღწერილობის ცხოველის საკვების ლოტის ნაწილიდან ნიმუშის აღებისას, თუ გამოვლინდება, რომ ლოტის ეს ნაწილი არ აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს, მაშინ ჩაითვლება რომ ამ ლოტის ცხოველის მთლიანი საკვები ისეთივეა და ლოტის დარჩენილი ნაწილიც არ აკმაყოფილებს აღნიშნულ მოთხოვნებს, გარდა იმ შემთხვევისა, როდესაც დეტალური შეფასების შემდეგ იქნება მოპოვებული მტკიცებულება იმის შესახებ, რომ დარჩენილი ნაწილი აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს. 8.4.2. სასილოსეში ნიმუშის აღება (დახურული სასილოსე), რომელთანაც წვდომა შეუძლებელია ზევიდან 8.4.2.1. ნიმუშის აღება სასილოსეში, რომელთანაც წვდომა შეუძლებელია ზევიდან (დახურული სასილოსე) > 100 ტონა. ასეთ სასილოსეში შენახული ცხოველის საკვების ნიმუშის აღება შეუძლებელია სტატიკური გზით. ამიტომ, თუ უნდა განხორციელდეს სასილოში ცხოველის საკვების ნიმუშის აღება და თუ არ არსებობს ტვირთის გადაადგილების შესაძლებლობა, ოპერატორთან უნდა შედგეს შეთანხმება, რომ მან უნდა შეატყობინოს ინსპექტორს, როდის მოხდება სასილოსეს გადმოტვირთვა, რათა შესაძლებელი იყოს ნიმუშის აღება ცხოველის საკვების ჩამოტვირთვის დროს. 8.4.2.2. ნიმუშის აღება სასილოსეში, რომელთანაც წვდომა შეუძლებელია ზევიდან (დახურული სასილოსე) < 1 0 0 ტონა. ნიმუშის აღების პროცედურა გულისხმობს 50-დან 100 კგ-მდე ჭურჭელში მოთავსებას და იქიდან ნიმუშის აღებას. გაერთიანებული ნიმუშის ზომა (სიდიდე) შეესაბამება მთლიანი ლოტის, ხოლო ინკრემენტალური ნიმუშების რაოდენობა ნიმუშის ასაღებად ჭურჭელში მოთავსებულ სილოსის რაოდენობას. ერთი და იმავე კლასის და აღწერილობის ცხოველის საკვების ლოტის ნაწილიდან ნიმუშის აღებისას, თუ გამოვლინდება, რომ ლოტის ეს ნაწილი არ აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს, მაშინ ჩაითვლება რომ ამ ლოტის ცხოველის მთლიანი საკვები ისეთივეა და ლოტის დარჩენილი ნაწილიც არ აკმაყოფილებს აღნიშნულ მოთხოვნებს, გარდა იმ შემთხვევისა, როდესაც დეტალური შეფასების შემდეგ იქნება მოპოვებული მტკიცებულება იმის შესახებ, რომ დარჩენილი ნაწილი აკმაყოფილებს საქართველოს კანონმდებლობით განსაზღვრულ მოთხოვნებს. 8.5. დიდ კონტეინერებში მოთავსებული ცხოველის შეუფუთავი საკვებიდან ნიმუშების აღება ასეთი ლოტებიდან ნიმუშების აღება უმეტესწილად შესაძლებელია მხოლოდ გადმოტვირთვის დროს. გარკვეულ შემთხვევებში შეუძლებელია გადმოტვირთვა იმპორტის ან კონტროლის ეტაპზე, და სწორედ ამის გამო, ნიმუშის აღება უნდა მოხდეს ასეთი კონტეინერების გადმოტვირთვისას. 9. ნიმუშების აღებასთან, მომზადებასა და შეფუთვასთან დაკავშირებული ინსტრუქციები 9.1. ზოგადი მოთხოვნა ნიმუშის აღება და მომზადება უნდა მოხდეს ზედმეტი დაყოვნების გარეშე, იმ სიფრთხილის ზომების გათვალისწინებით, რომელიც აუცილებელია იმის უზრუნველსაყოფად, რომ არ მოხდეს პროდუქტის შეცვლა და დაბინძურება. ნიმუშის ასაღებად განკუთვნილი ინსტრუმენტები, ზედაპირები და კონტეინერები, უნდა იყოს სუფთა და მშრალი. 9.2. ინკრემენტალური ნიმუში ინკრემენტალური ნიმუშები შემთხვევითი (რანდომული) შერჩევის პრინციპით აიღება შერჩეული პორციის მთლიანი რაოდენობიდან და ისინი უნდა იყოს დაახლოებით თანაბარი ზომის. ინკრემენტალური ნიმუშის ზომა სულ მცირე 100 გრამია ან უხეში საკვების ან ფურაჟის შემთხვევაში 25 გრამი დაბალი კუთრი წონით. იმ შემთხვევაში, თუ აღებული უნდა იქნეს 40-ზე ნაკლები ინკრიმენტალური ნიმუში, ამ დანართის მე-8 პუნქტით დადგენილი ნიმუშის აღების პროცედურების მიხედვით, ინკრიმენტული ნიმუშების ზომა (სიდიდე) განისაზღვრება გაერთიანებული ნიმუშის იმ საჭირო სიდიდის მიხედვით, რომლის მიღწევაც არის აუცილებელი (იხ. ამ დანართის პუნქტი 6). მცირე ლოტისგან შემდგარი შეფუთული ცხოველის საკვებიდან, ნიმუშის აღების შემთხვევაში, როდესაც რაოდენობრივი მოთხოვნების შესაბამისად აღებული უნდა იქნეს ინკრემენტალური ნიმუშების შეზღუდული რაოდენობა, ინკრემენტალური ნიმუში უნდა იყოს ერთი ორიგინალი ერთეულის შემცველობის, რომლის შემცველობა არ უნდა აღემატებოდეს 1 კგ-ს ან ერთ ლიტრს. მცირე ზომის ერთეულებისგან შემდგარი ცხოველის შეფუთული საკვების ნიმუშის აღების შემთხვევაში (მაგ. <250 გ), ინკრემენტალური ნიმუშის ზომა (სიდიდე) დამოკიდებულია ერთეულის ზომაზე. 9.2.1. ცხოველის შეუფუთავი საკვები საჭიროების შემთხვევაში, ნიმუშის აღება შეიძლება განხორციელდეს შერჩეული პორციის გადაადგილებისას (ჩატვირთვა ან გადმოტვირთვა). 9.2.2. ცხოველის შეფუთული საკვები ამ დანართის მე-5 პუნქტით გათვალისწინებული მოთხოვნების დაცვით, ნიმუშის ასაღებად საჭირო რაოდენობის ერთეულების შერჩევის შემდეგ, თითოეული ერთეულის შემცველობის ნაწილი უნდა მოსცილდეს შუბის ან ნიჩბის გამოყენებით. საჭიროების შემთხვევაში, ნიმუშების აღება ხდება ერთეულების ცალკე დაცლის შემდეგ. 9.2.3. ჰომოგენური ან ჰომოგენიზირებადი ცხოველის თხევადი ან ნახევრად თხევადი საკვები ამ დანართის მე-5 პუნქტით გათვალისწინებული მოთხოვნების დაცვით, ნიმუშის ასაღებად საჭირო რაოდენობის ერთეულების შერჩევის შემდეგ, შემცველობა, საჭიროების შემთხვევაში, ჰომოგენიზირდება და თითოეული ერთეულისგან აიღება რაოდენობა. ინკრემენტალური ნიმუშების აღება შესაძლებელია შემცველობის გადმოტვირთვის დროს. 9.2.4. არაჰომოგენიზირებადი, ცხოველის თხევადი ან ნახევრად თხევადი საკვები ამ დანართის მე-5 პუნქტით გათვალისწინებული მოთხოვნების დაცვით, ნიმუშის ასაღებად საჭირო რაოდენობის ერთეულების შერჩევის შემდეგ, ნიმუშები აღებული უნდა იქნეს სხვადასხვა დონიდან. ნიმუშების აღება ასევე შეიძლება შემცველობის გადმოტვირთვის დროსაც, მაგრამ პირველი ფრაქციები წუნდებულია (უგულველყოფილია). ნებისმიერ შემთხვევაში, არსებული ნიმუშის მთლიანი მოცულობა არ უნდა იყოს 10 ლიტრზე ნაკლები. 9.2.5. ცხოველის საკვები ბრიკეტები და სალოკი ამ დანართის მე-5 პუნქტით გათვალისწინებული მოთხოვნების დაცვით, ნიმუშის ასაღებად საჭირო რაოდენობის ბრიკეტებისა ან სალოკის შერჩევის შემდეგ, ხდება თითოეული ბრიკეტის ან სალოკის ნაწილის აღება. არაჰომოგენიზირებული ბრიკეტის ან სალოკის ეჭვის შემთხვევაში, მთლიანი ბრიკეტი ან სალოკი შეიძლება იქნეს აღებული ნიმუშად. არაუმეტეს 1 კგ წონის მქონე ბრიკეტის ან სალოკის შემთხვევაში, ინკრემენტალურ ნიმუშს წარმოადგენს ერთი ბრეკეტის ან ერთი სალოკის შემცველობა. 9.3. გაერთიანებული ნიმუშების მომზადება ერთი გაერთიანებული ნიმუშის შესაქმნელად შერეული უნდა იქნეს ინკრემენტალური ნიმუშები. 9.4. საბოლოო ნიმუშების მომზადება გაერთიანებული ნიმუშის მასალა გულდასმით უნდა იქნეს არეული1. თითოეული ნიმუში უნდა მოთავსდეს სათანადო კონტეინერში/ჭურჭელში/ტარაში. მიღებული უნდა იქნეს ყველა აუცილებელი სიფრთხილის ზომა, რათა თავიდან იქნეს აცილებული ნიმუშის შემადგენლობის რაიმე სახის ცვლილება, დაბინძურება ან გაფუჭება, რაც შეიძლება წარმოიშვას ტრანსპორტირების ან შენახვის დროს. კომპონენტების ან ნივთიერებების კონტროლის შემთხვევაში, რომლებიც თანაბრად ნაწილდება საკვებში, გაერთიანებული ნიმუში შეიძლება რეპრეზენტატიულად შემცირდეს სულ მცირე 2.0 კგ ან 2.0 ლიტრამდე (შემცირებული ნიმუში)2, სასურველია, მექანიკური ან ავტომატური გამყოფის გამოყენებით. პარკოსნებში, პარკოსან მარცვლეულებსა და კაკლოვანი კულტურების ნაყოფებში, პესტიციდების ნაშთი ოდენობის (ნარჩენების) არსებობის გასაკონტროლებლად შემცირებული ნიმუშის მინიმალური ზომა უნდა იყოს 3 კგ. იმ შემთხვევაში, თუ საკვების ბუნება არ იძლევა გამყოფის გამოყენების შესაძლებლობას ან გამყოფი არ არის ხელმისაწვდომი, მაშინ ნიმუში შეიძლება შემცირდეს კვარტირების მეთოდით. შემცირებული ნიმუშებიდან უნდა მომზადდეს საბოლოო ნიმუშები (კონტროლის, დაცვისა და რეფერენტული) დაახლოებით იმავე ოდენობით და ამ დანართის მე-7 პუნქტით განსაზღვრული რაოდენობრივი მოთხოვნების შესაბამისად. კომპონენტების, მათ შორის გენეტიკურად მოდიფიცირებული მასალის, ან ნივთიერებების კონტროლის შემთხვევაში, რომლებიც სავარაუდოდ, არაერთგვაროვნად ნაწილდება ცხოველის საკვებ მასალებში, გაერთიანებული ნიმუში უნდა იყოს: – მთლიანად ჰომოგენიზირებული და შემდეგ დაყოფილი საბოლოო ნიმუშებად; – ან შემცირებული სულ მცირე 2 კგ-ით ან 2 ლიტრით3 მექანიკური ან ავტომატური გამყოფის გამოყენებით. მხოლოდ იმ შემთხვევაში, თუ საკვების ბუნება არ იძლევა გამყოფის გამოყენების საშუალებას, საჭიროების შემთხვევაში, ნიმუში შეიძლება შემცირდეს კვარტირების მეთოდით. გენმოდიფიცირებული მასალის კონტროლისთვის, რომლისთვისაც ავტორიზაციის პროცედურა გადაწყვეტილების მოლოდინშია ან რომლის ავტორიზაციის ვადა ამოიწურა, შემცირებული ნიმუში უნდა შეიცავდეს არანაკლებ 35 000 თესლს/მარცვალს, რათა მოპოვებულ იქნეს სულ მცირე 10 000 თესლის/მარცვალის (იხ. ამ დანართის მე-6 პუნქტის სქოლიო (**) და მე-7 პუნქტის სქოლიო (*)) საბოლოო ნიმუში, კონტროლის, დაცვისა და რეფერენტული გამოკვლევისათვის. 9.5. ნიმუშების შეფუთვა კონტეინერების ან შეფუთვების/პაკეტების დალუქვა და ეტიკეტირება ისე უნდა მოხდეს, რომ შეუძლებელი იყოს მათი გახსნა ლუქის დაზიანების გარეშე. ეტიკეტი მთლიანად უნდა იყოს მოქცეული ლუქში. 9.6. ნიმუშების ლაბორატორიაში გაგზავნა ნიმუში, ანალიტიკოსისთვის საჭირო ინფორმაციასთან ერთად, იგზავნება წინასწარ განსაზღვრულ ლაბორატორიაში, ზედმეტი დაყოვნების გარეშე. 10. ნიმუშის აღების ოქმი თითოეული ნიმუშის აღებისას უნდა შეივსოს ოქმი, რათა მოხდეს თითოეული შერჩეული პორციისა და მისი ზომის (სიდიდის) ერთმნიშვნელოვანი იდენტიფიკაცია. ოქმში ასევე მითითებული უნდა იყოს ამ წესით გათვალისწინებული ნიმუშის აღების პროცედურიდან ნებისმიერი გადახრა/გადაცდომა. გარდა იმისა, რომ ოქმი დგება კონტროლის განმახორციელებელი ლაბორატორიისთვის, ოქმი ხელმისაწვდომი უნდა იყოს ცხოველის საკვების ბიზნესოპერატორის და/ან ცხოველის საკვების ბიზნესოპერატორის მიერ მითითებული ლაბორატორიისათვის. დანართი №2 ზოგადი დებულებები ცხოველის საკვების გამოკვლევის მეთოდების შესახებ თავი I ნიმუშების მომზადება გამოკვლევისთვის 1. მიზანი ამ თავში აღწერილი პროცედურები შეეხება ნიმუშების გამოკვლევისათვის მომზადებას, რომლებიც ამ წესის დანართი №1-ის შესაბამისად აღების შემდეგ იგზავნება კონტროლის განმახორციელებელ ლაბორატორიაში. ლაბორატორიული ნიმუშების მომზადება უნდა განხორციელდეს ისე, რომ გამოკვლევის მეთოდით განსაზღვრული ნიმუშების აწონილი რაოდენობა იყოს ერთგვაროვანი (ჰომოგენური) და რეპრეზენტატული საბოლოო ნიმუშებისთვის. 2. სიფრთხილის ზომები ნიმუშის მომზადების პროცედურა, დამოკიდებულია გამოკვლევის გამოსაყენებელ მეთოდებზე და გასაკონტროლებელ კომპონენტებზე ან ნივთიერებ ებზე. ამიტომ უდიდესი მნიშვნელობა აქვს იმაში დარწმუნებას, რომ ნიმუშის მომზადების პროცედურა შეესაბამებოდეს გამოკვლევის გამოყენებულ მეთოდსა და გასაკონტროლირებელ კომპონენტებს ან ნივთიერებებს. ყველა საჭირო ოპერაცია უნდა შესრულდეს ისე, რომ მაქსიმალურად იქნეს აცილებული თავიდან ნიმუშის დაბინძურება და მისი შემადგენლობის ცვლილება. დაფქვა, შერევა და გაცრა უნდა განხორციელდეს დაუყოვნებივ, სინჯის ჰაერზე და სინათლეზე მინიმალური ზემოქმედებით. ისეთი წისქვილები და საფქვავები, რომლებიც მნიშვნელოვნად აცხელებენ ნიმუშს არ უნდა იქნეს გამოყენებული. ხელით დაფქვა რეკომენდებულია ცხოველის საკვებისთვის, რომლებიც განსაკუთრებით მგრძნობიარეა სითბოს მიმართ. აგრეთვე, ყურადღება უნდა მიექცეს იმას, რომ თავად ხელსაწყო არ იყოს დაბინძურების წყარო. თუ მომზადება შეუძლებელია ნიმუშის სინესტის მნიშვნელოვანი ცვლილებების გარეშე, უნდა განისაზღვროს სინესტე მომზადებამდე და მის შემდეგ, ამ წესის დანართი №3-ის I თავით განსაზღვრული მეთოდის შესაბამისად. 3. პროცედურა 3.1. ზოგადი პროცედურა ალიქოტის ტესტი აიღება საბოლოო ნიმუშიდან. კონუსის ფორმირება და კვარტირება (ნიმუშის მომზადება ოთხად დაყოფის მეთოდით) რეკომენდებული არ არის, რადგან ამან შეიძლება გამოიწვიოს ალიქოტის ტესტის მაღალი ცდომილება გაყოფის თვალსაზრისით. 3.1.1. ცხოველის საკვები, რომელიც შეიძლება დაიფქვას აურიეთ გაცრილი საბოლოო ნიმუში და მოათავსეთ იგი სათანადო სუფთა, მშრალ მჭიდრო საცობიან ჭურჭელში. ზუსტად ნიმუშების გამოკვლევისათვის საჭირო რაოდენობის აწონვამდე, კვლავ აურიეთ სრული ჰომოგენიზაციის უზრუნველსაყოფად (ალიქოტის ტესტი). 3.1.2. ცხოველის საკვები, რომელიც შეიძლება დაიფქვას გაშრობის შემდეგ თუ გამოკვლევის მეთოდებში სხვა რამ არ არის გათვალისწინებული, საბოლოო ნიმუში გააშრეთ ისე, რომ მისი სინესტის შემცველობა შემცირდეს 8-დან 12%-მდე, იმ წინასწარი გაშრობის პროცედურის შესაბამისად, რომელიც აღწერილია სინესტის დადგენის მეთოდში (ამ წესის დანართი №3-ის I თავის 4.3 პუნქტი). შემდეგ პროცედურა გააგრძელეთ ისე, როგორც აღწერილია ამ თავის 3.1.1 პუნქტში. 3.1.3. ცხოველის თხევადი ან ნახევრად თხევადი საკვები შეაგროვეთ საბოლოო ნიმუში შესაბამის სუფთა და მშრალ ჭურჭელში, რომელსაც აქვს მჭიდრო საცობი. ზუსტად ნიმუშების გამოკვლევისათვის საჭირო რაოდენობის აწონვამდე, კვლავ საფუძვლიანად აურიეთ სრული ჰომოგენიზაციის უზრუნველსაყოფად (ალიქოტის ტესტი). 3.1.4. ცხოველის სხვა საკვები საბოლოო ნიმუშები, რომელთა მომზადება შეუძლებელია ზემოთ ჩამოთვლილი პროცედურების შესაბამისად, უნდა დამუშავდეს ნებისმიერი სხვა პროცედურით, რომელიც უზრუნველყოფს, გამოკვლევისთვის საჭირო აწონილი რაოდენობის (ალიქოტის ტესტი) ჰომოგენურობას და საბოლოო ნიმუშების რეპრეზენტატიულობას. 3.2. სპეციფიკური პროცედურა, რომელიც გამოიყენება ვიზუალური დათვალიერებით (ინსპექტირებით) ან მიკროსკოპით გამოკვლევისას ან იმ შემთხვევებში, როდესაც ხდება გაერთიანებული ნიმუშის ჰომოგენიზაცია – ვიზუალური დათვალიერებით (ინსპექტირებით) გამოკვლევის (მიკროსკოპის გამოყენების გარეშე) შემთხვევაში, გამოიყენება მთლიანი ლაბორატორიული ნიმუში. – მიკროსკოპული გამოკვლევის შემთხვევაში, ლაბორატორიამ შეიძლება შეამციროს გაერთიანებული ნიმუში, ან კიდევ უფრო შეამციროს შემცირებული ნიმუში. დაცვისათვის და საბოლოო რეფერენტული გამოკვლევის მიზნებისთვის განკუთვნილი საბოლოო ნიმუშების აღება ხდება პროცედურის დაცვით, რომელიც ეკვივალენტურია იმ პროცედურისა, რომელიც ხორციელდება საბოლოო ნიმუშის კონტროლისათვის. – მთლიანი გაერთიანებული ნიმუშის ჰომოგენიზაციის შემთხვევაში, საბოლოო ნიმუშების აღება ხდება ჰომოგენიზებული გაერთიანებული ნიმუშიდან. 4. ნიმუშების შენახვა ნიმუშები უნდა ინახებოდეს ისეთ ტემპერატურაზე, რომელიც არ გამოიწვევს მათი შემადგენლობის ცვლილებას. ვიტამინების ან ნივთიერებების, რომლებიც სინათლის მიმართ გამოირჩევიან განსაკუთრებული მგრძნობელობით, გამოკვლევისათვის განკუთვნილი ნიმუშები უნდა ინახებოდეს ისეთ პირობებში, რომ სინათლემ არ მოახდინოს ნიმუშზე უარყოფითი გავლენა. თავი II გამოკვლევის მეთოდებში გამოყენებული რეაგენტებსა და ხელსაწყოებთან დაკავშირებული მოთხოვნები 1. თუ გამოკვლევის მეთოდებში სხვა რამ არ არის მითითებული, ყველა ანალიტიკური რეაგენტი უნდა იყოს ანალიტიკურად სუფთა (ა.ს.). როდესაც ტარდება კვალის ანალიზი, რეაგენტების სიწმინდე უნდა შემოწმდეს საკონტროლო (ბრმა) ტესტით. მიღებული შედეგებიდან გამომდინარე, შეიძლება საჭირო გახდეს რეაგენტების დამატებითი გაწმენდა (პურიფიკაცია). 2. ნებისმიერი ოპერაცია, რომელიც მოიცავს ხსნარების მომზადებას, გაზავებას, ჩამორეცხვას ან რეცხვას, რაც აღნიშნულია გამოკვლევის მეთოდებში და რაზედაც არ არის არანაირი აღნიშვნა გამხსნელის ან გამზავებლის თვისებებზე, ნიშნავს იმას, რომ გამოყენებული უნდა იქნეს წყალი. ზოგადი წესის მიხედვით, წყალი უნდა იყოს დემინერალიზებული ან გამოხდილი. კონკრეტულ შემთხვევებში, რომლებიც მითითებულია გამოკვლევის მეთოდებში, იგი უნდა დაექვემდებაროს გაწმენდის (პურიფიკაციის) სპეციალურ პროცედურებს. 3. კონტროლის განმახორციელებელ ლაბორატორიებში გამოყენებული აღჭურვილობის გათვალისწინებით, გამოკვლევის მეთოდებში აღნიშნულია მხოლოდ ის ინსტრუმენტები და აპარატურა, რომლებიც სპეციალურია ან საჭიროებს სპეციფიკურ გამოყენებას. ისინი უნდა იყოს სუფთა, განსაკუთრებით მაშინ, როდესაც უნდა მოხდეს ძალიან მცირე რაოდენობის ნივთიერებების განსაზღვრა. თავი III გამოკვლევის მეთოდების გამოყენება და შედეგების წარდგენა 1. ექსტრაქციის პროცედურა ექსტრაქციის კონკრეტულ პროცედურას განსაზღვრავს რამდენიმე მეთოდი. ზოგადი წესის მიხედვით, მეთოდში ექსტრაქციაზე მითითებული პროცედურის გარდა, შეიძლება გამოყენებულ იქნეს ექსტრაქციის სხვა პროცედურები იმ პირობით, თუ მატრიცის გამოკვლევისთვის გამოყენებული ექსტრაქციის პროცედურით მტკიცდება იმ ეკვივალენტური ექსტრაქციის ეფექტურობა, რაც გათვალისწინებულია მეთოდში. 2. გაწმენდის პროცედურა გაწმენდის კონკრეტულ პროცედურას განსაზღვრავს რამდენიმე მეთოდი. ზოგადი წესის მიხედვით, მეთოდში მითითებული გაწმენდის პროცედურის გარდა, შეიძლება გამოყენებულ იქნეს გაწმენდის სხვა პროცედურები იმ პირობით, თუ მატრიცის გამოკვლევისთვის გამოყენებული გაწმენდის პროცედურის შედეგად მტკიცდება, იმ ეკვივალენტური გამოკვლევის შედეგების მიღწევა, რაც მეთოდშია გათვალისწინებული. 3. განსაზღვრის რაოდენობა გამოკვლევისას არასასურველი ნივთიერებების აღმოჩენის შემთხვევაში, თუ პირველი განსაზღვრის/გამოკვლევის შედეგი საკონტროლო სპეციფიკაციაზე მნიშვნელოვნად (> 50%) დაბალია, მაშინ დამატებითი განსაზღვრა/გამოკვლევა აღარ არის საჭირო, იმ პირობით, თუ გამოიყენება ხარისხის კონტროლის სათანადო პროცედურები. სხვა შემთხვევებში საჭიროა განმეორებითი (დუბლიკატი) განსაზღვრა/გამოკვლევა (მეორე განსაზღვრა), რათა გამოირიცხოს ნიმუშების შიდა ჯვარედინი დაბინძურება ან შემთხვევითი შერევა. შესაბამისობის ვერიფიკაციისათვის გამოიყენება ორი განსაზღვრის საშუალო მნიშვნელობა, განუსაზღვრელობის განსაზღვრის გათვალისწინებით. ნივთიერების ან ინგრედიენტის დეკლარირებული შემცველობის კონტროლის შემთხვევაში, თუ პირველი განსაზღვრის/გამოკვლევის შედეგი დაადასტურებს დეკლარირებულ შემცველობას, ე.ი. გამოკვლევის შედეგი დეკლარირებული შემცველობის ვარიაციის მისაღებ (დასაშვებ) დიაპაზონშია, დამატებითი განსაზღვრა/გამოკვლევა საჭირო აღარ არის, იმ პირობით, თუ გამოიყენება ხარისხის კონტროლის სათანადო პროცედურები. სხვა შემთხვევებში საჭიროა განემორებითი (დუბლიკატი) გამოკვლევა (მეორე განსაზღვრა), რომ გამოირიცხოს ნიმუშების შიდა ჯვარედინი დაბინძურება ან შემთხვევითი შერევა. შესაბამისობის ვერიფიკაციისათვის გამოიყენება ორი განსაზღვრის საშუალო მნიშვნელობა, განუსაზღვრელობის განსაზღვრის გათვალისწინებით. ზოგიერთ შემთხვევაში, ეს ვარიაციების მისაღები (დასაშვები) დიაპაზონი განისაზღვრება „ცხოველის საკვების გამოყენებისა და ბაზარზე განთავსების წესის დამტკიცების შესახებ“ საქართველოს მთავრობის 2021 წლის 8 თებერვალის №58 დადგენილებითა და „ცხოველთა კვებაში გამოსაყენებელი ცხოველის საკვები დანამატების წესის დამტკიცების შესახებ“ საქართველოს მთავრობის დადგენილებით. 4. გამოყენებული გამოკვლევის მეთოდის შესახებ ანგარიშგება გამოკვლევის ოქმში აღინიშნული უნდა იქნეს გამოყენებული გამოკვლევის მეთოდი. 5. გამოკვლევის შედეგების ოქმი გამოკვლევის შედეგი უნდა აისახოს ისე, როგორც ის დადგენილია გამოკვლევის მეთოდში, მნიშვნელოვანი მონაცემების შესაბამისი რაოდენობის გამოყენებით, და საჭიროების შემთხვევაში, მომზადებამდე უნდა მოხდეს საბოლოო ნიმუშის სინესტის შემცველობის კორექტირება. 6. განუსაზღვრელობის განსაზღვრა და აღდგენის მაჩვენებელი არასასურველი ნივთიერებების გამოკვლევის შემთხვევაში საქართველოს მთავარობის დადგენილებით განსაზღვრულ არასასურველ ნივთიერებებთან დაკავშირებით, ცხოველის საკვებად განკუთვნილი პროდუქტი უნდა ჩაითვალოს, რომ არ შეესაბამება დადგენილ მაქსიმალურ შემცველობას, როდესაც გამოკვლევის შედეგით, 12%-იანი სინესტის შემცველობის მქონე ცხოველის საკვებთან დაკავშირებით ითვლება, რომ აღემატება მაქსიმალურ შემცველობას, განუსაზღვრელობის გაფართოებული განსაზღვრისათვის და აღდგენისათვის საჭირო კორექციის გათვალისწინებით. შესაბამისობის შეფასებისთვის, გამოკვლეული კონცენტრაცია გამოიყენება მას შემდეგ, რაც მოხდება აღდგენისათვის საჭირო კორექტირება და განუსაზღვრელობის გაფართოებული განსაზღვრის გამორიცხვა. ეს პროცედურა გამოიყენება მხოლოდ იმ შემთხვევებში, როდესაც გამოკვლევის მეთოდი განუსაზღვრელობის განსაზღვრის შეფასებისა და აღდგენის კორექტირების საშუალებას იძლევა (მაგ.: მიკროსკოპული გამოკვლევის შემთხვევაში შეუძლებელია). ოქმში გამოკვლევის შედეგის შეტანა უნდა მოხდეს შემდეგნაირად (ამ დროისთვის გამოყენებული გამოკვლევის მეთოდი საშუალებას იძლევა შეფასდეს განუსაზღვრელობის განსაზღვრა და აღდგენის მაჩვენებელი): ა) კორექტირებულია აღდგენისთვის, მითითებულია აღდგენის დონე. კორექტირება აღდგენისათვის საჭირო არ არის იმ შემთხვევაში, თუ აღდგენის მაჩვენებელი 90-110%-ს შორისა; ბ) როგორც მაჩვენებლები „x +/– U“, სადაც x გამოკვლევის შედეგია და U განუსაზღვრელობის გაფართოებული განსაზღვრა, დაფარვის 2 კოეფიციენტის გამოყენებით, რომელიც იძლევა დაახლოებით 95% სანდოობას. მაგრამ, თუ გამოკვლევის შედეგი მნიშვნელოვნად (> 50%) დაბალია, სპეციფიკაციაზე, რომელიც ექვემდებარება კონტროლს, და იმ პირობით, რომ გამოიყენება შესაბამისი ხარისხის კონტროლის პროცედურები და გამოკვლევა ემსახურება მხოლოდ საკანონმდებლო მოთხოვნების შესაბამისობის შემოწმებას, გამოკვლევის შედეგი შეიძლება დაფიქსირდეს ოქმში აღდგენისათვის საჭირო კორექციის გარეშე, და ასეთ შემთხვევებში ოქმში აღდგენის მაჩვენებელი და განუსაზღვრელობის განსაზღვრა შეიძლება არ იქნეს მითითებული.
დანართი №3 ცხოველის საკვების მასალისა და კომბინირებული საკვების შემადგენლობის კონტროლისათვის გამოყენებული გამოკვლევის მეთოდები თავი I სინესტის განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას გვაძლევს განვსაზღვროთ ცხოველის საკვებში სინესტის შემცველობა. თუ ცხოველის საკვები შეიცავს ისეთ აქროლებად ნივთიერებებს, როგორიც არის ორგანული მჟავეები, აუცილებელია სინესტის შემცველობის განსაზღვრასთან ერთად აქროლებადი ნივთიერებების მნიშვნელოვანი რაოდენობის განსაზღვრაც. ეს მეთოდი არ მოიცავს რძის ნაწარმის გამოკვლევას, რომელიც გამოიყენება, როგორც ცხოველის საკვების მასალა, გამოკვლევას მინერალური ნივთიერებებსა და ნარევებზე, რომლებიც ძირითადად შედგება მინერალური ნივთიერებებისგან, გამოკვლევას ცხოველური და მცენარეული ცხიმებსა და ზეთებზე, ან გამოკვლევას ზეთოვანი კულტურების თესლსა და კაკლოვანების ნაყოფზე. 2. გამოყენების პრინციპები ნიმუშის გამოშრობა (დესიკაცია) წარმოებს გარკვეულ პირობებში, რომელიც განსხვავდება ცხოველის საკვების თვისებების მიხედვით. წონის დაკარგვა დგინდება აწონვით. როდესაც საქმე გვაქვს ცხოველის მაღალი სინესტის მქონე მყარ საკვებთან, აუცილებელია მისი წინასწარ გამოშრობა. 3. აპარატურა 3.1. დამაქუცმაცებელი აპარატი/დანადგარი, რომელიც დამზადებულია ისეთი მასალისაგან, რომელიც არ იწოვს სინესტეს, ადვილად იწმინდება, უზრუნველყოფს სწრაფ და თანაბრ დაფქვას შესამჩნევი გათბობის გარეშე, მაქსიმალურად ხელს უშლის გარე ჰაერთან კონტაქტს და შეესაბამება ამ თავის 4.1.1 და 4.1.2 პუნქტებით განსაზღვრულ მოთხოვნებს (მაგ., ჩაქუჩისებრი მიკროსამსხვრეველები ან მიკროსამსხვრეველები წყლის გამაგრილებლით, დრეკადი კონუსური სამსხვრეველები ან სამსხვრეველები შენელებული მოძრაობებით ან სამსხვრეველები დაკბილული დისკებით). 3.2. გამოსაკვლევად განკუთვნილი სასწორი 1 მგ სიზუსტით. 3.3. მშრალი ლითონის კონტეინერები, რომლებიც არ ექვემდებარება კოროზიას ან მინის კონტეინერები მჭიდრო სახურავებით; სამუშაო ზედაპირი, რომელიც ნიმუშის გაფანტვის საშუალებას იძლევა დაახლოებით 0.3 გ/სმ2 ფარგლებში. 3.4. იზოთერმული ღუმელი ელექტროგათბობით (± 2°C), სათანადო ვენტილაციით, რაც საშუალებას იძლევა ტემპერატურის სწრაფ დარეგულირებას (4). 3.5. რეგულირებადი ვაკუუმის ღუმელი ელექტროგამათბობელით, რომელსაც გააჩნია ზეთის ტუმბო და ცხელი მშრალი ჰაერის მიწოდების მექანიზმი ან საშრობი შემადგენლობა (მაგ., კალციუმის ოქსიდი). 3.6. საშრობი (დესიკატორი) სქელი პერფორირებული ლითონის ან ფაიფურის ფირფიტით, რომელიც შეიცავს ეფექტურ გამაშრობელ საშუალებას. 4. პროცედურა შენიშვნა: ამ ნაწილში აღწერილი ოპერაციები უნდა განხორციელდეს ნიმუშების შეფუთვების გახსნისთანავე. გამოკვლევა უნდა ჩატარდეს სულ მცირე განმეორებით (დუბლიკატ) ეგზემპლარად. 4.1. მომზადება 4.1.1. ცხოველის საკვები, რომელიც არ არის მითითებული ამ თავის 4.1.2 და 4.1.3 პუნქტებში აიღება სულ მცირე 50 გრ. ნიმუში. საჭიროების შემთხვევაში, იგი ისე უნდა დაიშალოს ან გაიყოს, რომ არ მოხდეს შემცველობაში სინესტის რაიმე ცვლილება (ვარიაცია)(იხ. ამ თავის პუნქტი 6). 4.1.2. ბურღულეული და ქატო აიღება სულ მცირე 50 გრ. ნიმუში. დაფქვით ნაწილაკებად, რომელთაგან სულ მცირე 50% გაივლის 0,5 მმ-იანი ხვრელების მქონე საცერში და დატოვებს არაუმეტეს 10% ნარჩენს 1 მმ-იან ოვალურ ხვრელიან საცერზე. 4.1.3. თხევადი ან პასტის ფორმის ცხოველის საკვები, ძირითადად ზეთებისა და ცხიმებისგან შემდგარი ცხოველის საკვები აიღეთ სულ მცირე 25 გრ. ნიმუში, აწონეთ 10 მგ სიზუსტით, დაამატეთ შესაბამისი რაოდენობის მშრალი ქვიშა, რომელიც აწონილი იქნა 10-მგ-იანი სიზუსტით, და ურიეთ მანამ სანამ არ მიიღებთ ჰომოგენურ პროდუქტს. 4.2. გაშრობა 4.2.1. ცხოველის საკვები, რომელიც არ არის მითითებული ამ თავის 4.2.2 და 4.2.3 პუნქტებში კონტეინერი (ამ თავის პუნქტი 3.3) აწონეთ თავსახურით 1 მგ-მდე სიზუსტით. აწონილ ჭურჭელში აწონეთ 1 მგ-მდე სიზუსტით 5 გრ-იანი ნიმუში და გაანაწილეთ თანაბრად. განათავსეთ კონტეინერი სახურავის გარეშე წინასწარ 103°C ტემპერატურაზე გახურებულ ღუმელში. ზედმეტად რომ არ მოხდეს ღუმელში ტემპერატურის ვარდნა, კონტეინერი მოათავსეთ ღუმელში, რაც შეიძლება სწრაფად. მას შემდეგ რაც ტემპერატურა დაუბრუნდება 103°C გრადუსს, გამოშრობას აცადეთ ოთხი საათი. კონტეინერზე თავსდება სახურავი, ხდება მისი ღუმელიდან გამოღება და 30-45 წუთის განმავლობაში გასაგრილებლად თავსდება საშრობში (დესიკატორი), (ამ თავის პუნქტი 3.6) და აწონეთ 1 მგ-ის სიზუსტით. ცხოველის საკვები, რომელიც უპირატესად შედგება ზეთებისა და ცხიმებისგან, გააშრეთ ღუმელში დამატებით 30 წუთის განმავლობაში, 130°C ტემპერატურაზე. ორ აწონას შორის სხვაობა არ უნდა აღემატებოდეს სინესტის 0,1%-ს. 4.2.2. მარცვლეული, ფქვილი, ბურღული და მსხვილად ნაფქვავი კონტეინერი (ამ თავის პუნქტი 3.3) აწონეთ თავსახურით 0.5 მგ-მდე სიზუსტით. აწონილ ჭურჭელში აწონეთ 1 მგ-მდე სიზუსტით 5 გრ-იანი დაქუცმაცებული ნიმუში და გაანაწილეთ თანაბრად. განათავსეთ კონტეინერი სახურავის გარეშე, წინასწარ 130°C ტემპერატურაზე გახურებულ ღუმელში. ზედმეტად რომ არ მოხდეს ღუმელში ტემპერატურის ვარდნა, კონტეინერი მოათავსეთ ღუმელში რაც შეიძლება სწრაფად. მას შემდეგ რაც ტემპერატურა დაუბრუნდება 130°C გრადუსს, გამოშრობას აცადეთ ორი საათი. კონტეინერზე თავსდება სახურავი, ხდება მისი ღუმელიდან გამოღება და 30-45 წუთის განმავლობაში გასაგრილებლად თავსდება საშრობში (ამ თავის პუნქტი 3.6) და ხდება მისი აწონა 1 მგ სიზუსტით. 4.2.3. ცხოველის კომბინირებული საკვები, რომელიც შეიცავს 4%-ზე მეტ საქაროზას ან ლაქტოზას: ცხოველის საკვების მასალები, როგორიცაა, ლობიო (locust beans), ჰიდროლიზებული ბურღულეულის პროდუქტები, ალაოს თესლი, გამხმარი ჭარხლის ჩიპსი, თევზისა და შაქრის პასტები; ცხოველის კომბინირებული საკვები, რომელიც შეიცავს მინერალური მარილების 25%-ზე მეტს, კრისტალიზებული წყლის ჩათვლით. კონტეინერი (ამ თავის პუნქტი 3.3.) აწონეთ თავსახურით 0.5 მგ-მდე სიზუსტით. აწონილ ჭურჭელში აწონეთ 1 მგ-მდე სიზუსტით 5 გრ-იანი ნიმუში და გაანაწილეთ თანაბრად. განათავსეთ კონტეინერი სახურავის გარეშე, წინასწარ 80-დან 85°C ტემპერატურაზე გახურებულ ვაკუუმის ღუმელში (ამ თავის პუნქტი 3.5). ზედმეტად რომ არ მოხდეს ღუმელში ტემპერატურის ვარდნა, კონტეინერი მოათავსეთ ღუმელში რაც შეიძლება სწრაფად. წნევა დააყენეთ 100 ტორამდე და დატოვეთ გასაშრობად ოთხი საათით ამავე წნევაზე, ან გააშრეთ ცხელი, მშრალი ჰაერის ნაკადით ან საშრობი საშუალების გამოყენებით (დაახლოებით 300გ 20 ნიმუშისთვის). ამ უკანასკნელ შემთხვევაში გათიშეთ ვაკუუმის ტუმბო, როდესაც მიიღწევა რეკომენდებული წნევა. დროის ათვლა დაიწყეთ იმ მომენტიდან, როდესაც ღუმელის ტემპერატურა დაუბრუნდება 80-დან 85°C გრადუსს. ფრთხილად დაიყვანეთ ღუმელი ატმოსფერულ წნევამდე. გააღეთ ღუმელი, დაუყოვნებლივ მოათავსეთ სახურავი კონტეინერზე, გამოიღეთ კონტეინერი ღუმელიდან, გააჩერეთ 30-45 წუთის განმავლობაში გაგრილებისთვის საშრობში (ამ თავის პუნქტი 3.6) და აწონეთ 1 მგ სიზუსტით. გააშრეთ ვაკუუმის ღუმელში დამატებით 30 წუთით 80-დან 85°C ტემპერატურაზე და ხელახლა აწონეთ. ორ აწონას შორის სხვაობა არ უნდა აღემატებოდეს სინესტის 0,1%-ს. 4.3. წინასწარი გაშრობა 4.3.1. ცხოველის საკვები, რომელიც არ არის მითითებული ამ თავის 4.3.2 პუნქტში მყარი საკვები სინესტის მაღალი შემცველობით, რომელიც ართულებს მის დაქუცმაცებას, უნდა დაექვემდებაროს წინასწარ გაშრობას, შემდეგნაირად: აწონეთ 50 გ დაუქუცმაცებელი ნიმუში 10 მგ-მდე სიზუსტით (კომპრესირებული ან აგლომერირებული ცხოველის საკვები შეიძლება უხეშად გაიყოს საჭიროების შემთხვევაში) შესაბამის კონტეინერში (მაგ. 20 × 12 სმ ალუმინის ფირფიტა 0,5 სმ-იანი ზღუდით). გასაშრობად გააჩერეთ ღუმელში 60-დან 70°C ტემპერატურაზე სანამ სინესტის შემცველობა არ დაიწევს 8%-დან 12%-მდე. გამოიღეთ ღუმელიდან, გასაგრილებლად თავმოხდილ მდგომარეობაში დატოვეთ ლაბორატორიაში ერთი საათის განმავლობაში და აწონეთ 10მგ-მდე სიზუსტით. დაუყოვნებლივ დააქუცმაცეთ ისე, როგორც ეს ამ თავის 4.1.1. პუნქტშია მითითებული და გააშრეთ ისე, როგორც მითითებულია ამ თავის 4.2.1 ან 4.2.3 პუნქტებში, ცხოველის საკვების თვისებებიდან გამომდინარე. 4.3.2. მარცვლეული 17%-ზე მეტი სინესტის შემცველობის მარცვლეული უნდა დაექვემდებაროს წინასწარ გაშრობას შემდეგნაირად: აწონეთ 50 გ დაუფქვავი მარცვალი 10 მგ სიზუსტით შესაბამის კონტეინერში (მაგ., 20 × 12 სმ ალუმინის ფირფიტა 0,5 სმ ზღუდით). გასაშრობად გააჩერეთ 5-7 წუთით 130°C ტემპერატურაზე ღუმელში. გამოიღეთ ღუმელიდან, თავმოხდილ მდგომარეობაში დატოვეთ ლაბორატორიაში ორი საათის განმავლობაში და აწონეთ 10 მგ-მდე სიზუსტით. დაუყოვნებლივ დააქუცმაცეთ ისე, როგორც ეს მითითებულია ამ თავის 4.1.2. პუნქტში და გააშრეთ ისე, როგორც მითითებულია ამ თავის 4.2.2 პუნქტში. 5. შედეგების დაანგარიშება სინესტის შემცველობა (X), როგორც ნიმუშის პროცენტული მაჩვენებელი, გამოითვლება შემდეგი ფორმულების გამოყენებით: 5.1. გაშრობა წინასწარი გაშრობის გარეშე
სადაც: m = გამოსაკვლევი ნიმუშის საწყისი წონა გრამებში; m0 = გამოსაკვლევი მშრალი ნიმუშის წონა გრამებში. 5.2. გაშრობა წინასწარი გაშრობით
სადაც: m = გამოსაკვლევი ნიმუშის საწყისი წონა გრამებში; m1 = გამოსაკვლევი ნიმუშის წონა გრამებში წინასწარი გაშრობის შემდეგ; m2 = გამოსაკვლევი ნიმუშის წონა გრამებში დაქუცმაცებისა და დაფქვის შემდეგ; m0 = გამოსაკვლევი მშრალი ნიმუშის წონა გრამებში. 5.3. განმეორებადობა ერთი და იმავე ნიმუშთან დაკავშირებით ორ პარალელურ განსაზღვრის შედეგებს შორის განსხვავება არ უნდა აღემატებოდეს სინესტის აბსოლუტური მნიშვნელობის 0,2 %-ს. 6. დაკვირვება თუ დაქუცმაცება აუცილებელი გახდება და თუ ეს გამოიწვევს ცვლილებას პროდუქტის სინესტის შემცველობაში, ცხოველის საკვების კომპონენტების გამოკვლევის შედეგები უნდა შესწორდეს ნიმუშის სინესტის საფუძველზე, ნიმუშის საწყისი მდგომარეობის გათვალისწინებით.
თავი II სინესტის განსაზღვრა ცხოველურ და მცენარეულ ცხიმებსა და ზეთებში 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას გვაძლევს განვსაზღვროთ წყლისა და აქროლადი ნივთიერებების შემცველობა ცხოველურ და მცენარეულ ცხიმებსა და ზეთებში. 2. გამოყენების პრინციპები ნიმუში შრება მუდმივი წონის მიღწევამდე (ორ თანმიმდევრულ აწონვებს შორის წონის კლება უნდა იყოს 1 მგ-ზე ნაკლები ან ტოლი) 103°C ტემპერატურაზე. წონის კლება დგინდება აწონვით. 3. აპარატურა 3.1. ბრტყელძირიანი ჭურჭელი, დამზადებულია კოროზიის მიმართ მდგრადი მასალისგან, არის დიამეტრით 8-დან 9 სმ-მდე და სიმაღლით დაახლოებით 3 სმ. 3.2. თერმომეტრი ფიქსირებული ბოლქვითა და ზედა ნაწილში გაფართოებული მილით, გრადირებული დაახლოებით 80°C-დან არანაკლებ 110°C-მდე და სიგრძით დაახლოებით 10სმ. 3.3. ქვიშის ავზი ან ელექტროფირფიტა. 3.4. საშრობი (დესიკატორი), რომელიც შეიცავს ეფექტურ გამაშრობელ რეაქტივს. 3.5. გამოსაკვლევად განკუთვნილი სასწორი. 4. მომზადების პროცედურა 1 მგ-მდე სიზუსტით აწონეთ 20გ ჰომოგენიზირებული ნიმუში მშრალ, აწონილ ჭურჭელში (ამ თავის პუნქტი 3.1), რომელიც შეიცავს თერმომეტრს (ამ თავის პუნქტი 3.2). ნიმუში გაათბეთ ქვიშის ავზში ან ცხელ ფირფიტაზე (ამ თავის პუნქტი 3.3), მოურიეთ თერმომეტრით განუწყვეტლივ ისე, რომ ტემპერატურამ მიაღწიოს 90°C გრადუსს დაახლოებით 7 წუთის განმავლობაში. შეამცირეთ სითბო, იმაზე დაკვირვებით, თუ რა სიხშირით ხდება ბუშტების ამოსვლა ჭურჭლის ფსკერიდან. ტემპერატურა არ უნდა აღემატებოდეს 105°C გრადუსს. გააგრძელეთ მორევა ჭურჭლის ფსკერის ზედაპირზე ხახუნით, სანამ ბუშტების წარმომშობა არ შეწყდება. სინესტის სრულად აღმოფხვრის უზრუნველსაყოფად, იგი რამდენჯერმე გააცხელეთ 103°C ± 2°C ტემპერატურაზე, თანმიმდევრულ გათბობებს შორის გაგრილებით 93°C-მდე. შემდეგ გააჩერეთ ოთახის ტემპერატურაზე გასაგრილებლად დესიკატორში (ამ თავის პუნქტი 3.4) და აწონეთ. ეს ოპერაცია გაიმეორეთ მანამ, სანამ წონის დაკლება ორ თანმიმდევრულ გათბობას შორის არ გადააჭარბებს 2 მგ-ს. შენიშვნა (Nota Bene: „კარგად დაუკვირდი“): ნიმუშის წონის ზრდა განმეორებითი გათბობის შემდეგ მიუთითებს ცხიმის დაჟანგვაზე, ამ შემთხვევაში დაიანგარიშეთ შედეგი იმ აწონიდან, რომელიც ჩატარდა უშუალოდ წონის გაზრდის დაწყებამდე. 5. შედეგების დაანგარიშება სინესტის შემცველობა (X), როგორც ნიმუშის პროცენტული მაჩვენებელი, გამოითვლება შემდეგი ფორმულის გამოყენებით:
სადაც: m = გამოსაკვლევი ნიმუშის საწყისი წონა გრამებში; m1 = ჭურჭლის წონა გრამებში თავისი შემცველობით გათბობამდე; m2 = ჭურჭლის წონაა გრამებში თავისი შემცველობით გათბობის შემდეგ. 0,05% -ზე დაბალი შედეგები უნდა დაფიქსირდეს, როგორც „0,05% -ზე დაბალი“ 6. განმეორებადობა ერთი და იმავე ნიმუშთან დაკავშირებით ორ პარალელურ განსაზღვრის შედეგებს შორის განსხვავება არ უნდა აღემატებოდეს სინესტის აბსოლუტური მნიშვნელობის 0,05 %-ს.
თავი III ნედლი პროტეინის შემცველობის განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას გვაძლევს განვსაზღვროთ ნედლი პროტეინის შემცველობა ცხოველის საკვებში აზოტის შემცველობის საფუზველზე, კელდალის მეთოდის გამოყენებით. 2. გამოყენების პრინციპი ნიმუში გადაიხარშება გოგირდმჟავაში კატალიზატორთან ერთად. მჟავას ხსნარი მზადდება ტუტე ნატრიუმის ჰიდროქსიდის ხსნარით. ამიაკის გამოხდა და შეგროვება ხდება გოგირდმჟავას გაზომილი რაოდენობით, რომლის ზედმეტობა იტიტრება ნატრიუმის ჰიდროქსიდის სტანდარტული ხსნარით. ალტერნატიულად, გამოთავისუფლებული ამიაკი იხსნება ბორის მჟავას ხნარის ჭარბი რაოდენობით და შემდეგ იტიტრება მარილმჟავას ან გოგირდმჟავას ხსნარით.
3. რეაგენტები 3.1. კალიუმის სულფატი. 3.2. კატალიზატორი: სპილენძის (II) ოქსიდი CuO ან სპილენძის (II) სულფატი პენტაჰიდრატი, CuSO4 5H2O. 3.3. გრანულირებული თუთია. 3.4. გოგირდმჟავა, ρ20 = 1,84 გ/მლ. 3.5. გოგირდმჟავა, სტანდარტული მოცულობითი ხსნარი, c (H2SO4) = 0,25 მოლი/ლიტრი. 3.6. გოგირდმჟავა, სტანდარტული მოცულობითი ხსნარი, c (H2SO4) = 0,10 მოლი/ლიტრი. 3.7. გოგირდმჟავა, სტანდარტული მოცულობითი ხსნარი, c (H2SO4) = 0,05 მოლი/ლიტრი. 3.8. მეთილის წითელის ინდიკატორი; გახსენით 300 მგ მეთილის წითელი 100 მლ ეთანოლში, σ = 95% -96% (მოც./მოც). 3.9. ნატრიუმის ჰიდროქსიდის ხსნარი (შეიძლება გამოყენებულ იქნეს ტექნიკური კლასი) β = 40 გ/100 მლ (მ/ვ: 40%). 3.10. ნატრიუმის ჰიდროქსიდის ხსნარი, სტანდარტული მოცულობითი ხსნარი c (NaOH) = 0,25 მოლი/ლიტრი. 3.11. ნატრიუმის ჰიდროქსიდი, სტანდარტული მოცულობითი ხსნარი c (NaOH) = 0,10 მოლი/ლიტრი. 3.12. გრანულირებული პემზის ქვა, გარეცხილი მარილმჟავით და კალცინირებული. 3.13. აცეტანილიდი (m.p. = 114 o C, N – შემცველობა = 10,36%). 3.14. საქაროზა (აზოტის გარეშე). 3.15. ბორის მჟავა (H3BO3). 3.16. მეთილის წითელის ინდიკატორის ხსნარი: გახსენით 100 მგ მეთილის წითელი 100 მლ ეთანოლში ან მეთანოლში. 3.17. ბრომოკრეზოლის მწვანე ხსნარი: გახსენით 100 მგ ბრომოკრეზოლი მწვანე 100 მლ ეთანოლში ან მეთანოლში. 3.18. ბორის მჟავას ხსნარი (10 გ/ლ-დან 40 გ/ლ-მდე იმის მიხედვით, თუ რომელი აპარატი/დანადგარი გამოიყენება). კოლორიმეტრული საბოლოო წერტილის გამოვლენის გამოყენებისას, ბორის მჟავას ხსნარში ემატება მეთილის წითელი და ბრომოკრეზოლის ინდიკატორი. თუ მომზადებულია 1 ლიტრი ბორის მჟავას ხსნარი, მოცულობის კორექტირებამდე უნდა დაემატოს 7 მლ მეთილის წითელის ინდიკატორის ხსნარი (ამ თავის პუნქტი 3.16) და 10 მლ ბრომოკრეზოლის მწვანე ხსნარი (ამ თავის პუნქტი 3.17). გამოყენებული წყლიდან გამომდინარე, ბორის მჟავას ხსნარის pH შეიძლება განსხვავდებოდეს პარტიების მიხედვით. მცირე რაოდენობით ტუტე ნივთიერებით კორექცია ხშირად საჭიროა დადებითი (ბრმა) კონტროლის მისაღებად. შენიშვნა: დაახლოებით 3 მლ-დან 4 მლ-მდე NaOH (ამ თავის პუნქტი 3.11) დამატება 1 ლიტრ 10 გ/ლ ბორის მჟავაში, როგორც წესი, იძლევა კარგ კორექტირებას. ხსნარი ინახება ოთახის ტემპერატურაზე და შენახვის დროს ხსნარი დაცული უნდა იყოს სინათლისა და ამიაკის ორთქლის წყაროებისგან. 3.19. მარილმჟავას სტანდარტული მოცულობითი ხსნარი c(HCl) = 0,10 მოლი/ლ. შენიშვნა: თუ გაანგარიშებებში ხორციელდება საჭირო კორექტირებები, შეიძლება გამოყენებულ იქნეს მოცულობითი ხსნარების სხვა კონცენტრაციები (ამ თავის პუნქტები 3.5, 3.6, 3.7, 3.10, 3.11 და 3.19). კონცენტრაციები ყოველთვის უნდა იყოს გამოხატული ოთხნიშნა ათწილადებით მძიმის შემდეგ. 4. აპარატურა შესაბამისი აპარატი/დანადგარი, რომელიც გამოიყენება გადახარშვის, დისტილაციისა და ტიტრაციისათვის კელდალის პროცედურის მიხედვით. 5. მომზადების პროცედურა 5.1. გადახარშვა აწონეთ 1 გ ნიმუში 0,001 გ-მდე სიზუსტით და ნიმუში გადაიტანეთ გადასახარშ აპარატის/დანადგარის კოლბაში. დაამატეთ 15გრ კალიუმის სულფატი (ამ თავის პუნქტი 3.1), შესაბამისი რაოდენობის კატალიზატორი (ამ თავის პუნქტი 3.2) (0,3-დან 0,4 გ სპილენძის (II) ოქსიდი ან 0,9-დან 1,2 გ სპილენძის (II) სულფატის პენტაჰიდრატი), 25 მლ გოგირდმჟავა (ამ თავის პუნქტი 3.4) და საჭიროების შემთხვევაში, რამდენიმე გრანულირებული პემზის ქვა (ამ თავის პუნქტი 3.12) და აურიეთ. თავიდან კოლბა შეათბეთ ზომიერად, საჭიროების შემთხვევაში დროდადრო ურიეთ წრიულად, ვიდრე მასა არ დანახშირდება და ქაფი არ გაქრება; შემდეგ უფრო ინტენსიურად გააცხელეთ, სანამ სითხე სტაბილურად არ დაიწყებს დუღილს. გათბობა ადეკვატურია, თუ მდუღარე მჟავა კონდენსირდება კოლბის კედელზე. არ დაუშვათ გვერდების გადახურება და ორგანული ნაწილაკების მასზე მიწებება. როდესაც ხსნარი გახდება გამჭვირვალე და ღია მწვანე ფერის, გააგრძელეთ დუღილი კიდევ ორი საათის განმავლობაში, შემდეგ დატოვეთ გასაგრილებლად. 5.2. დისტილაცია ფრთხილად დაამატეთ საკმარისი რაოდენობის წყალი სულფატების სრული დაშლის (გახსნის) უზრუნველსაყოფად. დააცადეთ გაგრილება და შემდეგ, საჭიროების შემთხვევაში, დაამატეთ თუთიის რამდენიმე გრანული (ამ თავის პუნქტი 3.3). გაგრძელეთ პროცედურა ამ თავის 5.2.1 ან 5.2.2 პუნქტების შესაბამისად. 5.2.1. დისტილაცია გოგირდმჟავაში დისტილაციის აპარატის/დანადგარის შემგროვებელ კოლბაში მოათავსეთ ზუსტად გაზომილი 25 მლ რაოდენობის გოგირდმჟავა (ამ თავის პუნქტი 3.5 ან 3.7), აზოტის სავარაუდო შემცველობის მიხედვით. დაამატეთ მეთილის წითელის ინდიკატორის რამდენიმე წვეთი (ამ თავის პუნქტი 3.8). გადასახარში აპარატის/დანადგარის კოლბა დააკავშირეთ დისტილაციის აპარატის/დანადგარის (დისტილიატორის) კონდენსატორთან და კონდენსატორის ბოლო ჩაუშვით სითხეში, რომელსაც შეიცავს შემგროვებელი კოლბა, სულ მცირე 1 სმ სიღრმეზე (იხ ამ თავის პუნქტი 8.3). ნელა ჩაასხით 100 მლ ნატრიუმის ჰიდროქსიდის ხსნარი (ამ თავის პუნქტი 3.9) გადასახარში აპარატის/დანადგარის კოლბაში ამიაკის დანაკარგის გარეშე (იხ. ამ თავის პუნქტი 8.1). გააცხელეთ კოლბა ვიდრე არ დასრულდება ამიაკის დისტილაცია. 5.2.2. დისტილაცია ბორის მჟავაში როდესაც დისტილატის ამიაკის შემცველობის ტიტრაცია ხდება ხელით, გამოიყენება ქვემოთ მოცემული პროცედურა. თუ დისტილაციის აპარატი/დანადგარი მთლიანად ავტომატიზირებულია და მოიცავს ამიაკის შემცველობის ტიტრაციას დისტილატში, მიჰყევით მწარმოებლის ინსტრუქციას დისტილაციის აპარატის/დანადგარის მოხმარების შესახებ. კონდენსატორის გამოსასვლელის ქვეშ მოათავსეთ შემგროვებელი კოლბა, რომელიც შეიცავს 25 მლ-დან 30 მლ-მდე ბორის მჟავას ხსნარს (ამ თავის პუნქტი 3.18), ისე რომ მიწოდების მილი (ტუბი) მოექცეს ბორის მჟავას ხსნარის მოჭარბებული ზედაპირის ქვემოთ. დისტილაციის აპარატი/დანადგარი დაარეგულირეთ ისე, რომ გამოთავისუფლდეს 50 მლ ნატრიუმის ჰიდროქსიდის ხსნარი (ამ თავის პუნქტი 3.9). გამოიყენეთ დისტილაციის აპარატი/დანადგარი მწარმოებლის ინსტრუქციის შესაბამისად და გამოხადეთ ამიაკი, რომელიც გამოიყოფა ნატრიუმის ჰიდროქსიდის ხსნარის დამატებით. შეაგროვეთ დისტილატი ბორის მჟავას შემცველ (მიმღებ) ხსნარში. დისტილატის რაოდენობა (ორთქლის გამოხდის დრო) დამოკიდებულია ნიმუშში არსებულ აზოტის ოდენობაზე. მიჰყევით მწარმოებლის მითითებებს. შენიშვნა: ნახევრად ავტომატური დისტილაციის აპარატში/დანადგარში ჭარბი ნატრიუმის ჰიდროქსიდის დამატება და ორთქლის გამოხდა ხორციელდება ავტომატურად. 5.3. ტიტრაცია იმოქმედეთ ამ თავის 5.3.1 ან 5.3.2 პუნქტის შესაბამისად. 5.3.1. გოგირდმჟავა მოახდინეთ ჭარბი გოგირდმჟავას ტიტრაცია შემგროვებელ კოლბაში ნატრიუმის ჰიდროქსიდის ხსნარით (ამ თავის პუნქტი 3.10 ან 3.11), გამოყენებული გოგირდმჟავას კონცენტრაციიდან გამომდინარე, საბოლოო წერტილის მიღწევამდე. 5.2.2. ბორის მჟავა მოახდინეთ შემგროვებელი კოლბის შემცველობის ტიტრაცია მარილმჟავას სტანდარტული მოცულობითი ხსნარით (ამ თავის პუნქტი 3.19) ან გოგირდმჟავას სტანდარტული მოცულობითი ხსნარით (ამ თავის პუნქტი 3.6) ბურეტის გამოყენებით და შეამოწმეთ (წაიკითხეთ) გამოყენებული ტიტრანტის რაოდენობა. როდესაც კოლორიმეტრული საბოლოო წერტილის გამოვლენა გამოიყენება, საბოლოო წერტილი მიიღწევა შემცველობაში პირველივე ვარდისფერი კვალის აღმოჩენისას. შეაფასეთ ბიურტის მაჩვენებელი 0,05 მლ სიზუსტით. განათებულმა მაგნიტურმა შემრევმა ფირფიტამ ან ფოტომეტრულმა დეტექტორმა შეიძლება ხელი შეუწყოს საბოლოო წერტილის ვიზუალიზაციას. ეს შეიძლება გაკეთდეს ავტომატურად, დისტილაციის აპარატის/დანადგარის (დისტილატორის) გამოყენებით, რომელსაც შეუძლია ავტომატური ტიტრაცია. მიჰყევით მწარმოებლის ინსტრუქციებს კონკრეტული დისტილატორის ან დისტილატორის/ტიტრატორის გამოყენების შესახებ. შენიშვნა: ტიტრაციის ავტომატური სისტემის გამოყენებისას, ტიტრაცია იწყება დისტილაციის დაწყებისთანავე და გამოიყენება ბორის მჟავას 1% -იანი ხსნარი (ამ თავის პუნქტი 3.18). როდესაც გამოიყენება მთლიანად ავტომატური დისტილაციის აპარატი/დანადგარი, ამიაკის ავტომატური ტიტრაცია ასევე შეიძლება განხორციელდეს საბოლოო წერტილის აღმოჩენისა და პოტენციომეტრიული pH სისტემის გამოყენებით. ამ შემთხვევაში გამოიყენება ავტომატური ტიტრატორი, pH-მეტრით. pH მრიცხველის სწორად დაკალიბრება უნდა მოხდეს pH4-დან pH 7-მდე დიაპაზონში, ნორმალური ლაბორატორიული pH საკალიბრო პროცედურების შესაბამისად. ტიტრაციის pH საბოლოო წერტილი მიიღწევა pH 4,6-ზე, რაც ტიტრაციის მრუდის ყველაზე დაბალ წერტილს წარმოადგენს (გადაღუნვის წერტილი). 5.4. ბრმა (საკონტროლო) ტესტი იმაში დასარწმუნებლად, რომ რეაგენტები არ შეიცავენ აზოტს, ჩაატარეთ ბრმა (საკონტროლო) ტესტი (გადახარშვა, დისტილაცია და ტიტრაცია) ნიმუშის ნაცვლად 1 გ საქაროზის გამოყენებით (ამ თავის პუნქტი 3.14). 6. შედეგების დაანგარიშება გაანგარიშება ხორციელდება ამ თავის 6.1 ან 6.2 პუნქტების მოთხოვნათა შესაბამისად. 6.1. ტიტრაციის დაანგარიშება ხორციელდება ამ თავის 5.3.1 პუნქტის შესაბამისად ნედლი პროტეინის შემცველობა, რომელიც პროცენტულად გამოხატულია წონაში, გამოითვლება შემდეგი ფორმულის მიხედვით:
სადაც: Vo = ბრმა (საკონტროლო) ტესტში გამოყენებული NaOH (ამ თავის 3.10 ან 3.11)-ის მოცულობა (მლ); V1 = ნიმუშის ტიტრაციაში გამოყენებული NaOH (ამ თავის პუნქტი 3.10 ან 3.11)-ის მოცულობა (მლ); c = ნატრიუმის ჰიდროქსიდის კონცენტრაცია (მოლი/ლიტრი) (ამ თავის პუნქტი 3.10 ან 3.11); m = ნიმუშის წონა (გ). 6.2. ტიტრაციის გაანგარიშება ხორციელდება ამ თავის 5.3.2 პუნქტის შესაბამისად 6.2.1. ტიტრაცია მარილმჟავასთან ნედლი პროტეინის შემცველობა, რომელიც პროცენტულად გამოხატულია წონაში, გამოითვლება შემდეგი ფორმულის მიხედვით:
სადაც: m = გამოსაკვლევი პორციის წონა (გ); c = მარილმჟავას სტანდარტული მოცულობითი ხსნარის კონცენტრაცია (მოლი/ლიტრი) (ამ თავის პუნქტი 3.19); V0 = ბრმა (საკონტროლო) ტესტისთვის გამოყენებული მარილმჟავას მოცულობა (მლ); V1 = გამოსაკვლევი პორციისათვის გამოყენებული მარილმჟავას მოცულობა (მლ). 6.2.2. ტიტრაცია გოგირდმჟავასთან ნედლი პროტეინის შემცველობა, რომელიც პროცენტულად გამოხატულია წონაში, გამოითვლება შემდეგი ფორმულის მიხედვით:
სადაც: m = გამოსაკვლევი პორციის წონა (გ); c = გოგირდმჟავას სტანდარტული მოცულობითი ხსნარის კონცენტრაცია (მოლი/ლიტრი) (ამ თავის პუნქტი 3.6); V0 = ბრმა (საკონტროლო) ტესტისთვის გამოყენებული გოგირდმჟავას მოცულობა (მლ) (ამ თავის პუნქტი 3.6); V1 = გამოსაკვლევი პორციისთვის გამოყენებული გოგირდმჟავას (მლ) მოცულობა (ამ თავის პუნქტი 3.6). 7. მეთოდის ვერიფიკაცია 7.1 განმეორებადობა ერთი და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – 0,2%-ს აბსოლუტური მნიშვნელობით, 20%-ზე ნაკლები ნედლი პროტეინის შემცველობისთვის; – 1%-ს უფრო მაღალ მნიშვნელობასთან მიმართებაში, ნედლი პროტეინის შემცველობისთვის 20-დან 40% -მდე; – 0,4%-ს აბსოლუტური მნიშვნელობით, 40%-ზე მეტი ნედლი პროტეინის შემცველობისთვის. 7.2. სიზუსტე ჩაატარეთ გამოკვლევა (გადახარშვა, დისტილაცია და ტიტრაცია) 1,5-დან 2,0 გ აცეტანილიდზე (ამ თავის პუნქტი 3.13) 1 გ საქაროზას (ამ თავის პუნქტი 3.14) არსებობისას; 1 გ აცეტანილიდი მოიხმარს 14,80 მლ გოგირდმჟავას (ამ თავის პუნქტი 3.5). აღდგენა უნდა იყოს არანაკლებ 99%. 8. დაკვირვება 8.1. აპარატი/დანადგარი შეიძლება იყოს მექანიკური, ნახევრად ავტომატური ან ავტომატური ტიპის. თუ აპარატი/დანადგარი საჭიროებს გადახარშვის და დისტილაციის საფეხურებს შორის გადართვას, ეს გადართვა უნდა განხორციელდეს დანაკარგის გარეშე. თუ დისტილაციის აპარატის/დანადგარის კოლბა არ არის აღჭურვილი წვეთოვანი ძაბრით (ჩამოსაშვები გვირაბით), მაშინ დაუმატეთ ნატრიუმის ჰიდროქსიდი უშუალოდ კოლბის კონდენსატორთან მიერთებამდე, თან სითხე ნელა ასხით ჭურჭლის კიდეზე. 8.2. თუ გადახარშული პროდუქტი მკვრივდება, განაახლეთ განსაზღვრა გოგირდმჟავას (ამ თავის პუნქტი 3.4) უფრო მეტი რაოდენობის გამოყენებით, როგორ ეს ზემოთ არის ნაჩვენები. 8.3. აზოტის დაბალი შემცველობის მქონე პროდუქტებზე, გოგირდმჟავას მოცულობა (ამ თავის პუნქტი 3.7), რომელიც უნდა მოთავსდეს შემგროვებელ კოლბაში, საჭიროების შემთხვევაში შეიძლება შემცირდეს 10 ან 15 მლ-მდე და შეადგინოს 25 მლ-მდე, წყალთან ერთად. 8.4. რუტინული გამოკვლევისთვის, ნედლი პროტეინის განსაზღვრისთვის, შეიძლება გამოყენებულ იქნეს გამოკვლევის ალტერნატიული მეთოდები, მაგრამ ამ თავში აღწერილი კელდალის მეთოდი არის რეფერენს მეთოდი. ალტერნატიული მეთოდით (მაგ.: DUMAS) მიღებული შედეგების ეკვივალენტობა რეფერენს მეთოდთან შედარებით უნდა იყოს ნაჩვენები თითოეული მატრიცისთვის ინდივიდუალურად. ვინაიდან ალტერნატიული მეთოდით მიღებულმა შედეგებმა, ეკვივალენტურობის გადამოწმების შემდეგაც, შეიძლება ოდნავ გამოიწვიოს ცდომილება რეფერენს მეთოდით მიღებულ შედეგებთან მიმართებაში, საჭიროა გამოკვლევის ოქმში აღინიშნოს ნედლი პროტეინის განსაზღვრისთვის გამოყენებული გამოკვლევის მეთოდი.
თავი IV შარდოვანას განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ შარდოვანას შემცველობა ცხოველის საკვებში. 2. გამოყენების პრინციპი ნიმუში გამწმენდი საშუალებით (აგენტით) სუსპენზირდება წყალში. სუსპენზია იფილტრება. ფილტრატის შარდოვანას შემცველობა განისაზღვრება 4-დიმეთილამინობენზალდეჰიდის (4-DMAB) დამატების შემდეგ ოპტიკური სიმკვრივის გაზომვით 420 ნმ ტალღის სიგრძეზე. 3. რეაგენტები 3.1. 4-დიმეთილამინობენზალდეჰიდის ხსნარი: გახსენით 1,6 გ 4-DMAB 100 მლ 96% ეთანოლში და დაამატეთ 10 მლ მარილმჟავა (ρ20 1,19 გ/მლ). ეს რეაგენტი ინახება არაუმეტეს ორი კვირის განმავლობაში. 3.2. Carrez-ის ხსნარი I: წყალში გახსენით 21,9 გ თუთიის აცეტატი, Zn (CH3 COO)2 2H2O და 3 გ დაკრისტალებული ძმარმჟავა. წყალით შეავსეთ 100 მლ-მდე. 3.3. Carrez-ის ხსნარი II: წყალში გახსენით 10,6 გ კალიუმის ფეროციანიდი, K4Fe(CN)6 3H2O. წყალით შეავსეთ 100 მლ-მდე. 3.4. აქტიური ნახშირბადი, რომელიც არ შთანთქავს შარდოვანას (უნდა შემოწმდეს). 3.5. შარდოვანა, 0,1% ხსნარი (წონ/მოც). 4. აპარატურა 4.1. მიქსერი (ტუმბლერი): დაახლოებით 35-დან 40-მდე ბრ/წთ. 4.2. სინჯარა: 160 × 16 მმ მინის გლუვზედაპირიანი საცობებით. 4.3. სპექტროფოტომეტრი. 5. გამოყენების პროცედურა 5.1. ნიმუშების გამოკვლევა აწონეთ 2 გ ნიმუში 1 მგ-ის სიზუსტით და 1 გ აქტიურ ნახშირბადთან ერთად მოათავსეთ (ამ თავის პუნქტი 3.4) 500 მლ მოცულობის კოლბაში. დაამატეთ 400 მლ წყალი და 5 მლ Carrez-ის ხსნარი I (ამ თავის პუნქტი 3.2), აურიეთ დაახლოებით 30 წამი და დაამატეთ 5 მლ Carrez-ის ხსნარი II (ამ თავის პუნქტი 3.3). ურიეთ 30 წუთის განმავლობაში მიქსერში (ტუმბლერში). საჭირო მოცულობის მისაღებად შეავსეთ წყლით, შეანჯღრიეთ და გაფილტრეთ. მოაცილეთ 5მლ გამჭვირვალე უფერული ფილტრატები, მოათავსეთ გამოსაკვლევ სინჯარაში, რომელსაც აქვს მინის გლუვზედაპირიანი საცობები, დაამატეთ 5 მლ 4-DMAB ხსნარი (ამ თავის პუნქტი 3.1) და აურიეთ. სინჯარები მოათავსეთ წყლის აბაზანაში 20oC (+/ – 4oC). 15 წუთის შემდეგ გაზომეთ ნიმუშის ხსნარის ოპტიკური სიმკვრივე სპექტროფოტომეტრით 420 ნმ-ზე. შეადარეთ რეაგენტების ხსნარი ბრმა (საკონტროლო) ტესტით. 5.2. საკალიბრო მრუდი მოაშორეთ 1, 2, 4, 5-ის მოცულობა და 10 მლ შარდოვანას ხსნარი (ამ თავის პუნქტი 3.5), მოათავსეთ 100 მლ მოცულობით კოლბებში და შეავსეთ მოცულობა წყლით. მოაშორეთ 5 მლ თითოეული ხსნარიდან, თითოეულს დაუმატეთ 5 მლ 4-DMAB ხსნარი (ამ თავის პუნქტი 3.1), მოახდინეთ ჰომოგენიზაცია და გაზომეთ ოპტიკური სიმკვრივე, როგორც ეს ნაჩვენებია ზემოთ, საკონტროლო ხსნართან შედარებით, რომელიც შეიცავს 5 მლ 4-DMAB და 5 მლ ისეთ წყალს, რომელიც არ შეიცავს შარდოვანას. დახაზეთ საკალიბრო მრუდი. 6. შედეგების დაანგარიშება განსაზღვრეთ შარდოვანას რაოდენობა ნიმუშში საკალიბრო მრუდის გამოყენებით. გამოხატეთ შედეგი, როგორც ნიმუშის პროცენტული მაჩვენებელი. 7. დაკვირვება 7.1. იმ შემთხვევაში, თუ შარდოვანას შემცველობა აღემატება 3%-ს, შეამცირეთ ნიმუში 1 გ-მდე ან შეაზავეთ ორიგინალი ხსნარი ისე, რომ 500 მლ-ში არ იყოს 50 მგ-ზე მეტი შარდოვანა. 7.2. შარდოვანას დაბალი შემცველობის შემთხვევაში, გაზარდეთ ნიმუში მანამ, სანამ ფილტრატი რჩება გამჭვირვალე და უფერო. 7.3. თუ ნიმუში შეიცავს მარტივ აზოტოვან ნაერთებს, როგორიცაა ამინომჟავები, ოპტიკური სიმკვრივე უნდა გაიზომოს 435 ნმ-ზე.
თავი V აქროლადი აზოტოვანი ფუძეების განსაზღვრა ქვეთავი I აქროლადი აზოტოვანი ფუძეების განსაზღვრა მიკროდიფუზიით 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ ცხოველის საკვებში ამიაკის სახით გამოხატული აქროლადი აზოტოვანი ფუძეების შემცველობა. 2. გამოყენების პრინციპი ნიმუშის ექსტრაჰირება ხდება წყლით, ხოლო ხსნარი იწმინდება და იფილტრება. აქროლადი აზოტოვანი ფუძეები გამოიდევნება მიკროდიფუზიით კალიუმის კარბონატის ხსნარის გამოყენებით, გროვდება ბორის მჟავას ხსნარში და იტიტრება გოგირდმჟავით. 3. რეაგენტები 3.1. ტრიქლორძმარ მჟავას ხსნარი 20% (წონ./მოც.). 3.2. ინდიკატორი: გახსენით 33 მგ ბრომოკრეზოლის მწვანე და 65 მგ მეთილის წითელი 100 მლ 95% -96% (მოც./მოც.)-ის ეთანოლში. 3.3. ბორის მჟავას ხსნარი: 1-ლიტრიან გრადირებულ კოლბაში გახსენით 10 გ ბორის მჟავა 200 მლ 95% -96% (მოც/მოც) ეთანოლში და 700 მლ წყალში. დაამატეთ 10 მლ ინდიკატორი (ამ ქვეთავის პუნქტი 3.2). შეურიეთ და საჭიროების შემთხვევაში, დააკორექტირეთ ხსნარის ფერი ღია წითელამდე, ნატრიუმის ჰიდროქსიდის ხსნარის დამატებით. აღნიშნული ხსნარის 1 მლ აფიქსირებს მაქსიმუმ 300 მგ NH3. 3.4. გაჯერებული კალიუმის კარბონატის ხსნარი: 100 მ კალიუმის კარბონატი გახსენით 100 მლ მდუღარე წყალში. დატოვეთ გასაგრილებლად და გაფილტრეთ. 3.5. გოგირდმჟავა 0,01 მოლი/ლიტრი. 4. აპარატურა 4.1. მიქსერი (ტუმბლერი): დაახლოებით 35-დან 40-მდე ბრ/წთ 4.2. მინის ან პლასტმასის კონვეის სექცია (იხ. დიაგრამა №1). 4.3. 1/100 მლ-ზე გრადირებული მიკრობურეტები. 5. მომზადების პროცედურა აწონეთ 10 გ ნიმუში 1 მგ-მდე სიზუსტით და 100 მლ წყალთან ერთად მოათავსეთ 200-მლ-იან მოცულობის გრადირებულ კოლბაში. აურიეთ ან შეანჯღრიეთ მიქსერში (ტუმბლერში) 30 წუთის განმავლობაში. დაამატეთ 50 მლ ტრიქლორძმარმჟავას ხსნარი (ამ ქვეთავის პუნქტი 3.1), წყლით შეავსეთ შესაბამის მოცულობამდე, კარგად შეანჯღრიეთ და გაფილტრეთ გოფრირებული ფილტრის საშუალებით. პიპეტის გამოყენებით შეიტანეთ 1 მლ ბორის მჟავას ხსნარი (ამ ქვეთავის პუნქტი 3.3) კონვეის სექციის ცენტრალურ ნაწილში და 1 მლ ფილტრატის ნიმუში სექციის თავში (გვირგვინში). ნაწილობრივ დაფარეთ ცხიმწასმული სახურავით. ჩააწვეთეთ სწრაფად გვირგვინში 1 მლ კალიუმის კარბონატის გაჯერებული ხსნარი (ამ ქვეთავის პუნქტი 3.4) და დახურეთ სახურავი ისე, რომ სექცია ჰერმეტულად იყოს დახურული. სექცია გადაატრიალეთ ფრთხილად ჰორიზონტალურ მდგომარეობაში, ისე რომ ორი რეაგენტი ერთმანეთში შეერიოს. დატოვეთ ინკუბაციისთვის, სულ მცირე, ან ოთხი საათის განმავლობაში ოთახის ტემპერატურაზე ან ერთი საათის განმავლობაში 40 oC ტემპერატურაზე. მიკრობურეტის გამოყენებით (ამ ქვეთავის პუნქტი 4.3), გოგირდმჟავით მოახდინეთ ბორის მჟავას ხსნარში აქროლადი ფუძეების ტიტრაცია (გატიტვრა) (ამ ქვეთავის პუნქტი 3.5). ჩაატარეთ ბრმა (საკონტროლო) ტესტი იმავე პროცედურის გამოყენებით, მაგრამ გამოსაკვლევი ნიმუშის გარეშე. 6. შედეგების ინტერპრეტაცია 1 მლ H2SO4 0,01 მოლი/ლიტრი შეესაბამება 0,34 მგ ამიაკს. გამოხატეთ შედეგი, როგორც ნიმუშის პროცენტული მაჩვენებელი. განმეორებადობა – ერთი და იმავე ნიმუშთან დაკავშირებით ორ პარალელურ განსაზღვრის შედეგებს შორის განსხვავება არ უნდა აღემატებოდეს: – 10%-ს ფარდობით მნიშვნელობაში, ამიაკის 1.0%-ზე ნაკლებ შემცველობისას; – 0,1%-ს აბსოლუტურ მნიშვნელობაში, ამიაკის 1,0%-ის ან მეტი შემცველობისას. 7. დაკვირვება თუ ნიმუშის ამიაკის შემცველობა აღემატება 0.6% -ს, გააზავეთ საწყისი ფილტრატი.
დიაგრამა №1 კონვეის სექცია მაშტაბი 1/1
ქვეთავი II აქროლადი აზოტოვანი ფუძეების განსაზღვრა დისტილაციით
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა დავადგინოთ ამიაკის სახით გამოხატული აქროლადი აზოტოვანი ფუძეების შემცველობა თევზის ფქვილში, რომელიც პრაქტიკულად არ შეიცავს შარდოვანას. იგი გამოიყენება მხოლოდ ამიაკის იმ შემცველობაზე, რომელიც 0,25%-ზე ნაკლებია. 2. გამოყენების პრინციპი ნიმუშის ექსტრაჰირება ხდება წყლით, ხოლო ხსნარი იწმინდება და იფილტრება. აქროლადი აზოტოვანი ფუძეების გამოდენა ხდება დუღილის მიღწევით მაგნიუმის ოქსიდის დამატებით და გროვდება გოგირდმჟავას განსაზღვრულ რაოდენობაში, რომლის ზედმეტობა იტიტრება ნატრიუმის ჰიდროქსიდის ხსნარით. 3. რეაგენტები 3.1. ტრიქლორძმარ მჟავას ხსნარი 20%, (წონ./მოც.) 3.2. მაგნიუმის ოქსიდი. 3.3. აქაფების საწინააღმდეგო ემულსია (მაგ., სილიკონი). 3.4. გოგირდმჟავა 0,05 მოლი/ლიტრი. 3.5. 0,1 მოლი/ლიტრი ნატრიუმის ჰიდროქსიდის ხსნარი. 3.6. მეთილის წითელი ხსნარი 0,3% 95% -96%-იან (მოცულობა/მოცულობა) ეთანოლში. 4. აპარატურა 4.1. მიქსერი (ტუმბლერი): დაახლოებით 35-დან 40-მდე ბრ/წთ. 4.2. კელდალის ტიპის დისტილაციის აპარატი/დანადგარი. 5. მომზადების პროცედურა აწონეთ 10 გ ნიმუში 1 მგ-მდე სიზუსტით და 100 მლ წყალთან ერთად მოათავსეთ 200-მლ-იან მოცულობის გრადირებულ კოლბაში. აურიეთ ან შეანჯღრიეთ მიქსერში (ტუმბლერში) 30 წუთის განმავლობაში. დაამატეთ 50 მლ ტრიქლოროძმარმჟავას ხსნარი (ამ ქვეთავის პუნქტი 3.1), წყლით შეავსეთ შესაბამის მოცულობამდე, კარგად შეანჯღრიეთ და გაფილტრეთ გოფრირებული ფილტრის საშუალებით. აიღეთ გამჭვირვალე ფილტრატის შესაბამისი რაოდენობა სავარაუდო აქროლადი აზოტოვანი ფუძეების შემცველობისთვის (როგორც წესი 100 მლ საკმარისია). გააზავეთ 200 მლ-მდე და დაამატეთ 2 გ მაგნიუმის ოქსიდი (ამ ქვეთავის პუნქტი 3.2) და რამდენიმე წვეთი აქაფების საწინააღმდეგო ემულსია (ამ ქვეთავის პუნქტი 3.3). ხსნარი უნდა იყოს ტუტე ლაკმუსის ქაღალდისთვის; წინააღმდეგ შემთხვევაში დაამატეთ მაგნიუმის ოქსიდი (ამ ქვეთავის პუნქტი 3.2). იმოქმედეთ ნედლი პროტეინის შემცველობის განსაზღვრისათვის ამ ქვეთავის 5.2 და 5.3 პუნქტების განსაზღვრული გამოკვლევის მეთოდის შესაბამისად (ამ დანართის თავი III). ჩაატარეთ ბრმა (საკონტროლო) ტესტი იმავე პროცედურის გამოყენებით, მაგრამ გამოსაკვლევი ნიმუშის გარეშე. 6. შედეგების ინტერპრეტაცია 1 მლ H2SO4 0,05 მოლი/ლიტრი შეესაბამება 1,7 მგ ამიაკს. გამოხატეთ შედეგი, როგორც ნიმუშის პროცენტული მაჩვენებელი.
განმეორებადობა – ერთი და იმავე ნიმუშზე შესრულებული ორ პარალელურ განსაზღვრის შედეგებს შორის სხვაობა, შედარებით მნიშვნელობაში, არ უნდა აღემატებოდეს ამიაკის 10 %-ს.
თავი VI ამინომჟავების განსაზღვრა (გარდა ტრიფტოფანისა) 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა, ამინომჟავების ანალიზატორის გამოყენებით, დავადგინოთ თავისუფალი (სინთეზური და ბუნებრივი) და საერთო (პეპტიდებთან შეკავშირებული და თავისუფალი) ამინომჟავები ცხოველის საკვებში. იგი გამოიყენება შემდეგ ამინომჟავებზე: ცისტ(ე)ინი, მეთიონინი, ლიზინი, ტრეონინი, ალანინი, არგინინი, ასპარტინის მჟავა, გლუტამინის მჟავა, გლიცინი, ჰისტიდინი, იზოლეიცინი, ლეიცინი, ფენილალანინი, პროლინი, სერინი, თიროზინი და ვალინი. ეს მეთოდი არ განასხვავებს ამინომჟავების მარილებს და მას არ შეუძლია განასხვაოს ამინომჟავების D და L ფორმები. ეს არ გამოიყენება ტრიფტოფანის ან ამინომჟავების ჰიდროქსი ანალოგების დასადგენად. 2. გამოყენების პრინციპი 2.1. თავისუფალი ამინომჟავები თავისუფალი ამინომჟავების ექსრაქტირება ხდება განზავებული მარილმჟავათი. ერთდროულად ექსტრაჰირებული აზოტოვანი მაკრომოლეკულა ილექება სულფოსალიცილის მჟავით და განცალკევდება ფილტრაციით. გაფილტრული ხსნარი დაიყვანება pH-ის 2,20 მნიშვნელობაზე. ამინომჟავები განცალკევდებიან იონგაცვლითი ქრომატოგრაფიით და განისაზღვრება ნინჰიდრინთან რეაქციით, ფოტომეტრული დეტექციით 570 ნმ-ზე. 2.2. ამინომჟავების საერთო შემცველობა პროცედურის შერჩევა დამოკიდებულია საკვლევ ამინომჟავაზე. ჰიდროლიზის დაწყებამდე, ცისტ(ე)ინი და მეთიონინი უნდა დაიჟანგოს ცისტეინის მჟავათი და შესაბამისად, მეთიონინის სულფონით. თიროზინი უნდა განისაზღვროს დაუჟანგავი ნიმუშების ჰიდროლიზატებში. ყველა სხვა ამინომჟავა, რომელიც ჩამოთვლილია ამ თავის პირველ პუნქტში, შეიძლება განისაზღვროს, ან დაჟანგულ, ან დაუჟანგავ ნიმუშში. დაჟანგვა ტარდება 0oC ტემპერატურაზე წარმოებული მჟავა/ფენოლის ნარევის მეშვეობით. ზედმეტი დაჟანგვის რეაგენტი იშლება ნატრიუმის დისულფიტით. დაჟანგული ან დაუჟანგავი ნიმუში ჰიდროლიზდება მარილმჟავით (ამ თავის პუნქტი 3.20) 23 საათის განმავლობაში. ჰიდროლიზატი დაიყვანება pH-ის 2,20 მნიშვნელობაზე. ამინომჟავები განცავლკევდებიან იონების გაცვლითი ქრომატოგრაფიით და განისაზღვრება ნინჰიდრინთან რეაქციით 570 ნმ-ზე ფოტომეტრული დეტექციის გამოყენებით (პროლინისთვის 440 ნმ). 3. რეაგენტები გამოყენებული უნდა იქნეს ორმაგი გამოხდილი წყალი ან ეკვივალენტური ხარისხის წყალი (გამტარობა <10 მკ S). 3.1. წყალბადის ზეჟანგი, წ (წ/წ) = 30%. 3.2. ფორმმჟავა(ჭიანჭველმჟავა), წ (წ/წ) = 98% – 100%. 3.3. ფენოლი. 3.4. ნატრიუმის დისულფიტი. 3.5. ნატრიუმის ჰიდროქსიდი. 3.6. 5-სულფოსალიცილის მჟავის დიჰიდრატი. 3.7. მარილმჟავა, დაახლოებით 1,18 გ/მლ. სიმკვრივით. 3.8. ტრი-ნატრიუმის ციტრატის დიჰიდრატი. 3.9. 2,2'-თიოდიეთანოლი (თიოდიგლიკოლი). 3.10. ნატრიუმის ქლორიდი. 3.11. ნინჰიდრინი. 3.12. პეტროლეუმის ეთერი, დუღილის დიაპაზონი 40-60oC. 3.13. ნორლეიცინი, ან სხვა ნაერთი, რომელიც ვარგისია შიდა სტანდარტის გამოსაყენებლად. 3.14. აზოტის გაზი (<10 ppm ჟანგბადი). 3.15. 1-ოქტანოლი. 3.16. ამინომჟავები. 3.16.1. ამ თავის 1.1. პუნქტში მოცემული სტანდარტული ნივთიერებები. სუფთა ნაერთები, რომლებიც არ შეიცავს კრისტალიზებულ წყალს. გამოყენებამდე 1 კვირით ადრე გამოაშრეთ ვაკუუმში P2O5 ან H2 SO4-ზე. 3.16.2. ცისტეინის მჟავა. 3.16.3. მეთიონინ სულფონი. 3.17. ნატრიუმის ჰიდროქსიდის ხსნარი, c = 7,5 მოლი/ლ: გახსენით 300 გ NaOH (3.5) წყალში და შეავსეთ 1 ლიტრამდე. 3.18. ნატრიუმის ჰიდროქსიდის ხსნარი, c = 1 მოლი/ლ: გახსენით 40 გ NaOH (ამ თავის პუნქტი 3.5) წყალში და შეავსეთ 1 ლიტრამდე. 3.19. ფორმმჟავა (ჭიანჭველმჟავა) – ფენოლის ხსნარი. შეურიეთ 889 გ ფორმმჟავა(ჭიანჭველმჟავა), (ამ თავის პუნქტი 3.2) 111 გ წყალში და დაუმატეთ 4.73 გ ფენოლი (ამ თავის პუნქტი 3.3). 3.20. ჰიდროლიზის ნარევი, c = 6 მოლი HCl/ლ, რომელიც შეიცავს 1 გ ფენოლს/ლ: დაამატეთ 1 გ ფენოლი (ამ თავის პუნქტი 3.3) 492 მლ HCl (ამ თავის პუნქტი 3.7)-ზე და წყლით შეავსეთ 1 ლიტრამდე. 3.21. ექსტრაქციის ნარევი, c = 0,1 მოლი HCl/ლ, რომელიც შეიცავს 2% თიოდიგლიკოლს: აიღეთ 8,2 მლ HCl (ამ თავის პუნქტი 3.7), გააზავეთ დაახლოებით 900 მლ წყლით, დაამატეთ 20 მლ თიოდიგლიკოლი (ამ თავის პუნქტი 3.9) და წყალით შეავსეთ 1 ლიტრამდე (ამ თავის პუნქტი 3.7 და 3.9 -ით განსაზღვრული ნივთიერებები პირდაპირ არ აურიოთ). 3.22. 5-სულფოსალიცილის მჟავა, ß = 6%: წყალში გახსენით 60 გ 5-სულფოსალიცილის მჟავა (ამ თავის პუნქტი 3.6) და შეავსეთ 1 ლ-მდე წყლით. 3.23. დამჟანგვი ნარევი (წარმოებული მჟავა – ფენოლი): მცირე ზომის მენზურაში შეურიეთ 0,5 მლ წყალბადის ზეჟანგი (ამ თავის პუნქტი 3.1) 4,5 მლ ფორმმჟავა (ჭიანჭველმჟავა) ფენოლის ხსნართან (ამ თავის პუნქტი 3.19). მოახდინეთ ინკუბაცია 20-30°C ტემპერატურაზე 1 საათის განმავლობაში, წარმოებული მჟავას წარმოქმნის მიზნით, შემდეგ გააგრილეთ ყინულის წყლის აბაზანაში (15 წთ.) ნიმუშის დამატებამდე. გაფრთხილება: მოერიდეთ კანთან კონტაქტს და ჩაიცვით დამცავი ტანსაცმელი. 3.24. ბუფერული ციტრატის ხსნარი, c = 0.2 მოლი Na +/ლ, pH 2.20: გახსენით 19.61 გ ნატრიუმის ციტრატი (ამ თავის პუნქტი 3.8), 5 მლ თიოდიგლიკოლი (ამ თავის პუნქტი 3.9), 1 გ ფენოლი (ამ თავის პუნქტი 3.3) და 16.50 მლ HCl (ამ თავის პუნქტი 3.7) დაახლოებით 800 მლ წყალში. pH დაიყვანეთ 2.20-ზე. წყლით შეავსეთ 1 ლიტრამდე. 3.25. ელუციის ბუფერები, დამზადებულია გამოყენებული ანალიზატორისათვის განკუთვნილი პირობების შესაბამისად (ამ თავის პუნქტი 4.9). 3.26. ნინჰიდრინის რეაგენტი, დამზადებულია გამოყენებული ანალიზატორისათვის განკუთვნილი პირობების შესაბამისად (ამ თავის პუნქტი 4.9). 3.27. ამინომჟავების სტანდარტული ხსნარები. ეს ხსნარები ინახება 5oC-ზე ნაკლებ ტემპერატურაზე. 3.27.1. ამინომჟავების საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.16.1). c = 2.5 მმოლი/მლ-ს თითოეულისთვის მარილმჟავაში. შეიძლება მიღებულ იქნეს კომერციული წყაროებიდან. 3.27.2. ცისტეინის მჟავას და მეთიონინის სულფონის საწყისი სტანდარტული ხსნარი, c = 1,25 მმოლი/მლ. 1-ლიტრიან გრადირებულ კოლბაში გახსენით 0,2115 გ ცისტეინის მჟავა (ამ თავის პუნქტი 3.16.2) და 0,2265 გ მეთიონინის სულფონი (ამ თავის პუნქტი 3.16.3) ციტრატის ბუფერში (ამ თავის პუნქტი 3.24) და შეავსეთ ციტრატის ბუფერით. შეინახეთ 5oC-ზე დაბალ ტემპერატურაზე არაუმეტეს 12 თვის განმავლობაში. ეს ხსნარი არ გამოიყენება, თუ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.27.1) შეიცავს ცისტეინის მჟავას და მეთიონინის სულფონს. 3.27.3. შიდა სტანდარტის საწყისი სტანდარტული ხსნარი მაგ. ნორლეუცინი, c = 20 მმოლი/მლ. გახსენით 0,6560 გ ნორლეუცინი (ამ თავის პუნქტი 3.13) ციტრატის ბუფერში (ამ თავის პუნქტი 3.24) გრადირებულ კოლბაში და შეავსეთ 250 მლ-მდე ციტრატის ბუფერით. შეინახეთ 5oC ტემპერატურაზე არაუმეტეს 6 თვის განმავლობაში. 3.27.4. ამინომჟავების საკალიბრო სტანდარტული ხსნარი, ჰიდროლიზატებთან გამოყენებისთვის, c = 5 ნმოლი/50 მკ l ცისტეინის მჟავით და მეთიონინის სულფონით, და c = 10 ნმოლი/50 მკ l-ით სხვა ამინომჟავებსთვის. გახსენით 2,2 გ ნატრიუმის ქლორიდი (ამ თავის პუნქტი 3.10) 100 მლ მენზურაში 30 მლ ციტრატის ბუფერთან (ამ თავის პუნქტი 3.24). დაამატეთ 4,00 მლ ამინომჟავების საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.27.1), 4,00 მლ ცისტეინის მჟავას და მეთიონინის სულფონის საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.27.2) და 0,50 მლ შიდა სტანდარტის საწყისი სტანდარტული ხსნარი (3.27.3) საჭიროების შემთხვევაში. pH მიიყვანეთ 2,20-მდე ნატრიუმის ჰიდროქსიდით (ამ თავის პუნქტი 3.18). რაოდენობრივად გადაიტანეთ 50 მლ გრადირებულ კოლბაში და ციტრატის ბუფერით შეავსეთ ნიშნულამდე (ამ თავის პუნქტი 3.24) და აურიეთ. შეინახეთ 5oC-ზე ნაკლებ ტემპერატურაზე არაუმეტეს 3 თვის განმავლობაში. აგრეთვე იხილეთ ამ თავის პუნქტი 9.1 – დაკვირვება. 3.27.5. ამინომჟავების საკალიბრო სტანდარტული ხსნარი ჰიდროლიზატებთან გამოსაყენებლად მომზადებულია ამ თავის 5.3.3.1 პუნქტის შესაბამისად და ექსტრაქტებთან (5.2) გამოსაყენებლად. საკალიბრო ხსნარი მზადდება ამ თავის 3.27.4 პუნქტის შესაბამისად, მაგრამ გამორიცხავს ნატრიუმის ქლორიდს. შეინახეთ 5oC-ზე დაბალ ტემპერატურაზე არაუმეტეს 3 თვის განმავლობაში. 4. აპარატურა 4.1. 100 ან 250 მლ მრგვალძირიანი კოლბა, რომელსაც აქვს რეფლუქსური კონდენსატორი. 4.2. 100 მლ ბოროსილიკატური მინის ბოთლი ხრახნიანი თავსახურით რეზინის/ტეფლონის სადებით (მაგ.: Duran, Schott) ღუმელში გამოსაყენებლად. 4.3. ღუმელი იძულებითი ვენტილაციით და ტემპერატურის მარეგულირებელი ± 2oC-ზე მეტი სიზუსტით. 4.4. pH მეტრი (სამნიშნა ათწილადებით მძიმის შემდეგ). 4.5. მემბრანული ფილტრი (0,22 მკმ). 4.6. ცენტრიფუგა. 4.7. მბრუნავი ვაკუუმის ამაორთქლებელი. 4.8. მექანიკური სადღვები ან მაგნიტური სათქვეფელა. 4.9. ამინომჟავების ანალიზატორი ან HPLC აპარატურა იონ-გაცვლითი სვეტით, მოწყობილობა ნინჰიდრინისთვის, პოსტ-სვეტის დერივატიზაცია და ფოტომეტრიული დეტექტორი. სვეტი ივსება სულფონირებული პოლისტიროლის ფისებით, რომლებსაც შეუძლიათ ამინომჟავების ერთმანეთისგან და სხვა ნინჰიდრინ-დადებითი მასალებისგან განცალკევება. ბუფერის და ნინჰიდრინის ხაზებში ნაკადი უზრუნველყოფილია ტუმბოებით, რომელთა ნაკადის სტაბილურობაა ± 0,5% იმ პერიოდში, რომელიც მოიცავს, როგორც სტანდარტულ საკალიბრო პროცესს, ისე ნიმუშის გამოკვლევას. ზოგიერთი ამინომჟავის ანალიზატორი შეიძლება გამოყენებულ იქნეს ჰიდროლიზის პროცედურებში, რომელშიც ჰიდროლიზატს აქვს ნატრიუმის კონცენტრაცია c = 0,8 მოლი/ლ და შეიცავს დაჟანგვის საფეხურიდან ფორმმჟავას (ჭიანჭველმჟავა) ნაშთ ოდენობას (ნარჩენს). სხვები არ იძლევიან ზოგიერთი ამინომჟავის დამაკმაყოფილებელ გამიჯვნას, თუ ჰიდროლიზატი შეიცავს ჭარბი ფორმმჟავას(ჭიანველმჟავა) და/ან ნატრიუმის იონის მაღალ კონცენტრაციებს. ამ შემთხვევაში მჟავას მოცულობა მცირდება დაახლეობით 5 მლ-ის აორთქლებით ჰიდროლიზის შემდეგ და pH-ის კორექტირებამდე. აორთქლება უნდა ჩატარდეს ვაკუუმში მაქსიმუმ 40oC ტემპერატურაზე. 5. გამოყენების პროცედურა 5.1. ნიმუშის მომზადება ნიმუში ქუცმაცდება და ტარდება 0.5 მმ-იანი ხვრების მქონე საცერში. მაღალი ტენიანობის შემცველი ნიმუშები უნდა გაშრეს ან ჰაერის ნაკადით არაუმეტეს 50oC ტემპერატურაზე, ან მოხდეს მათი მშრალად გაყინვა დაფქვის წინ. მაღალი რაოდენობის ცხიმშემცველი ნიმუშების ექსტრაჰირება ხდება პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.12) დაქუცმაცებამდე. 5.2. თავისუფალი ამინომჟავების განსაზღვრა ცხოველის საკვებსა და პრემიქსებში აწონეთ 0.2 მგ სიზუსტით შესაბამისი რაოდენობით (1-5 გ) მომზადებული ნიმუში (ამ თვაის პუნქტი 5.1), კონუსურ კოლბაში და დაამატეთ 100.0 მლ ექსტრაქტის ნარევი (ამ თავის პუნქტი 3.21). ანჯღრიეთ ნარევი 60 წუთი, მექანიკური სადღვების ან მაგნიტური სათქვეფელას გამოყენებით (ამ თავის პუნქტი 4.8). მიეცით ნალექს დაწმენდის (დალექვის) საშუალება და 10,0 მლ ნალექზედა ხსნარის შრე (სუპერნატანტი) პიპეტით გადაიტანეთ 100 მლ მენზურაში. დაამატეთ 5,0 მლ სულფოსალიცილის მჟავას ხსნარი (ამ თავის პუნქტი 3.22) მორევით და განაგრძეთ მორევა მაგნიტური სათქვეფელას დახმარებით 5 წუთის განმავლობაში. მოახდინეთ სუპერნატანტის გაფილტვრა ან ცენტრიფუგირება ნებისმიერი სახის ნალექის მოსაშორებლად. 10,0 მლ მიღებული ხსნარი მოათავსეთ 100 მლ მენზურაში და pH დააკორექტურეთ 2,20-მდე, ნატრიუმის ჰიდროქსიდის ხსნარის გამოყენებით (ამ თავის პუნქტი 3.18), გადაიტანეთ შესაბამისი მოცულობის კოლბაში ციტრატის ბუფერის გამოყენებით (ამ თავის პუნქტი 3.24), და ბუფერის ხსნარით შეავსეთ ნიშნულამდე (ამ თავის პუნქტი 3.24). შიდა სტანდარტის გამოყენებისას ემატება 1 მლ შიდა სტანდარტი (ამ ტავის პუნქტი 3.27.3) ყოველ 100 მლ საბოლოო ხსნარზე და ბუფერული ხსნარით შეავსეთ ნიშნულამდე (ამ თავის პუნქტი 3.24). გადადით ქრომატოგრაფიის ეტაპზე ამ თავის 5.4 პუნქტის შესაბამისად. თუ ექსტრაქტები არ მოწმდება იმავე დღეს, ისინი შენახული უნდა იქნან 5oC-ზე ნაკლებ ტემპერატურაზე. 5.3. ამინომჟავების საერთო შემცველობის განსაზღვრა 5.3.1. დაჟანგვა აწონეთ 0,2 მგ სიზუსტით 0,1-დან 1 გ-მდე მომზადებული ნიმუში (ამ თავის პუნქტი 5.1): – 100 მლ მრგვალძირიან კოლბაში (ამ თავის პუნქტი 4.1) ღია ჰიდროლიზისთვის (ღია გარემოში) (ამ თავის პუნქტი 5.3.2.3); – ან 250 მლ მრგვალძირიან კოლბაში (ამ თავის პუნქტი 4.1), თუ მოთხოვნილია ნატრიუმის დაბალი კონცენტრაცია (ამ თავის პუნქტი 5.3.3.1); – ან 100 მლ ბოთლში, რომელსაც აქვს ხრახნიანი თავსახური (ამ თავის პუნქტი 4.2), დახურული ჰიდროლიზისთვის (დახურულ გარემოში) (ამ თავის პუნქტი 5.3.2.4). აწონილი ნიმუშის პორციას უნდა ჰქონდეს აზოტის შემცველობა დაახლოებით 10 მგ და ტენიანობის შემცველობა არაუმეტეს 100 მგ-ისა. მოათავსეთ კოლბა/ბოთლი ყინულის წყლის აბაზანაში და გააგრილეთ 0oC ტემპერატურაზე, დაამატეთ 5 მლ დამჟანგავი ნარევი (ამ თავის პუნქტი 3.23) და აურიეთ მოხრილი წვერის მქონე მინის შპატელის გამოყენებით. ჰერმეტული ქსოვილით დალუქეთ კოლბა/ბოთლი, რომელიც შეიცავს შპატელს, მოათავსეთ ყინულის წყლის აბაზანაში, რომელიც შეიცავს დალუქულ ჭურჭელს, მაცივარში 0°C ტემპერატურაზე და გააჩერეთ 16 საათის განმავლობაში. 16 საათის შემდეგ გამოიღეთ მაცივრიდან და დაშალეთ ჭარბი დაჟანგული რეაგენტი 0,84 გ ნატრიუმის დისულფიტის დამატებით (ამ თავის პუნქტი 3.4). გადადით ამ თავის 5.3.2.1 პუნქტზე 5.3.2. ჰიდროლიზი 5.3.2.1. დაჟანგული ნიმუშების ჰიდროლიზი ამ თავის 5.3.1 პუნქტის შესაბამისად მომზადებულ დაჟანგულ ნიმუშს დაამატეთ 25 მლ ჰიდროლიზის ნარევი (ამ თავის პუნქტი 3.20), და იზრუნეთ, რომ ჭურჭლის და შპატელის გვერდებზე მიმაგრებული ნიმუშის ნაშთი (ნარჩენები) ჩამორეცხოთ. გამოყენებული ჰიდროლიზის პროცედურებიდან გამომიდინარე, გადადით ამ თავის 5.3.2.3 ან 5.3.2.4. პუნქტებში მითითებულ მეთოდებზე. 5.3.2.2. დაუჟანგავი ნიმუშების ჰიდროლიზი 2 მგ სიზუსტით აწონეთ 0,1-დან 1 გ-მდე მზა ნიმუში (ამ თავის პუნქტი 5.1), 100 მლ ან 250 მლ მრგვალძირიან კოლბაში (ამ თავის პუნქტი 4.1) ან 100 მლ ბოთლში, რომელსაც გააჩნია ხრახნიანი თავსახური (ამ თავის პუნქტი 4.2). აწონილი ნიმუშის პორციას უნდა გააჩნდეს დაახლოებით 10 მგ. აზოტის შემცველობა. ფრთხილად დაამატეთ 25 მლ ჰიდროლიზის ნარევი (ამ თავის პუნქტი 3.20) და შეურიეთ ნიმუშს. შემდეგ გააგრძელეთ ამ თავის 5.3.2.3 ან 5.3.2.4 პუნქტის შესაბამისად. 5.3.2.3. ღია ჰიდროლიზი (ჰიდროლიზი ღია გარემოში) კოლბაში არსებულ ნარევს დაუმატეთ 3 მინის ბურთულა (რომელიც მომზადებულია ამ თავის 5.3.2.1. ან 5.3.2.2. პუნქტის შესაბამისად) და ხარშეთ უწყვეტად სუსტი დუღილის რეჟიმით (ბუშტის გენერირებით, რეფლუქსის ქვეშ) 23 საათის განმავლობაში. ჰიდროლიზის დასრულების შემდეგ, ჩამორეცხეთ კონდენსატორი 5 მლ ციტრატის ბუფერით (ამ თავის პუნქტი 3.24). ჩახსენით კოლბა და გააგრილეთ იგი ყინულის აბაზანაში. შემდეგ გაგრძელეთ ამ თავის 5.3.3 პუნქტის შესაბამისად. 5.3.2.4. დახურული ჰიდროლიზი (ჰიდროლიზი დახურულ გარემოში) ბოთლი, რომელიც შეიცავს 5.3.2.1 ან 5.3.2.2 პუნქტის შესაბამისად მომზადებულ ნარევს, განათავსეთ ღუმელში (ამ თავის პუნქტი 4.3) 110oC ტემპერატურაზე. პირველი საათის განმავლობაში, წნევის დაგროვებისა (აიროვანი ნივთიერებების გამოყოფის გამო) და აფეთქების თავიდან აცილების მიზნით, ჭურჭლის თავზე მოათავსეთ ხრახნიანი თავსახური. არ დაახუროთ ჭურჭელს თავსახურით. დახურეთ ჭურჭელი თავსახურით ერთი საათის შემდეგ და გააჩერეთ ღუმელში (ამ თავის პუნქტი 4.3) 23 საათის განმავლობაში. ჰიდროლიზის დასრულების შემდეგ, ბოთლი ამოიღეთ ღუმელიდან, ფრთხილად გახსენით ბოთლის თავსახური და განათავსეთ ბოთლი ყინულიანი წყლის აბაზანაში. დატოვეთ გასაგრილებლად. pH-ის რეგულირების პროცედურიიდან გამომდინარე (ამ თავის პუნქტი 5.3.3), ბოთლის შემცველობა გადაიტანეთ რაოდენობრივად 250 მლ მენზურაში ან 250 მლ მრგვალძირიან კოლბაში, ციტრატის ბუფერის გამოყენებით (ამ თავის პუნქტი 3.24). შემდეგ გაგრძელეთ ამ თავის 5.3.3 პუნქტის შესაბამისად. 5.3.3. pH -ის რეგულირება ამინომჟავების ანალიზატორის ნატრიუმის ტოლერანტობიდან გამომდინარე (ამ თავის პუნქტი 4.9), pH-ის რეგულირების მიზნით იმოქმედეთ ამ თავის 5.3.3.1 ან 5.3.3.2 პუნქტში აღწერილი მეთოდის შესაბამისად. 5.3.3.1. pH-ის რეგულირება ქრომატოგრაფიული სისტემებისთვის (ამ თავის პუნქტი 4.9), რომლებიც საჭიროებენ ნატრიუმის დაბალ კონცენტრაციას ამინომჟავების ანალიზატორების გამოყენებისას, რომლებიც საჭიროებენ ნატრიუმის დაბალ კონცენტრაციას (როდესაც საჭიროა მჟავას მოცულობის შემცირება), სასურველია შიდა სტანდარტის საწყისი სტანდარტული ხსნარის გამოყენება (ამ თავის პუნქტი 3.27.3). ამ შემთხვევაში ჰიდროლიზატს აორთქლების წინ დაუმატეთ 2,00 მლ შიდა სტანდარტის საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.27.3). ამ თავის 5.3.2.3 ან 5.3.2.4 პუნქტის შესაბამისად მიღებულ ჰიდროლიზატს დაუმატეთ 1-ოქტანოლის (ამ თავის პუნქტი 3.15) 2 წვეთი. მბრუნავი ამაორთქლებლის გამოყენებით (ამ თავის პუნქტი 4.7) შეამცირეთ მოცულობა ვაკუუმში 5-10 მლ-მდე 40oC ტემპერატურაზე. თუ მოცულობა შემთხვევით შემცირდა 5 მლ-ზე ქვევით, ჰიდროლიზატი წუნდებული უნდა იქნეს და გამოკვლევა დაწყებული უნდა იქნეს ახლიდან. PH კორექტირდება 2,20-მდე ნატრიუმის ჰიდროქსიდის ხსნარით (ამ თავის პუნქტი 3.18) და გადადით ამ თავის 5.3.4 პუნქტზე. 5.3.3.2. PH-ის რეგულირება ყველა სხვა ამინომჟავების ანალიზატორებისთვის (ამ თავის პუნქტი 4. 9) აიღეთ ამ თავის 5.3.2.3 ან 5.3.2.4 პუნქტის შესაბამისად მიღებული ჰიდროლიზატები და ნაწილობრივ გაანეიტრალეთ 17 მლ ნატრიუმის ჰიდროქსიდის ხსნარის (ამ თავის პუნქტი 3.17) ფრთხილი დამატებით და თან ურიეთ, თან მიაქციეთ ყურადღება, რომ შენარჩუნებული იყოს 40oC-ზე დაბალი ტემპერატურა. pH დააკორექტირეთ 2,20-მდე ოთახის ტემპერატურაზე ნატრიუმის ჰიდროქსიდის ხსნარისა (ამ თავის პუნქტი 3.17) და საბოლოო ნატრიუმის ჰიდროქსიდის ხსნარის (ამ თავის პუნქტი 3.18) გამოყენებით. შემდეგ გადადით ამ თავის 5.3.4 პუნქტზე. 5.3.4. ნიმუშის ხსნარი ქრომატოგრაფიისთვის რაოდენობრივად გადაიტანეთ pH-ით კორექტირებული ჰიდროლიზატი (ამ თავის პუნქტი 5.3.3.1. ან 5.3.3.2.) ციტრატის ბუფერით (ამ თავის პუნქტი 3.24) 200-მლ-იან გრადირებულ კოლბაში და ბუფერული ხსნარით შეავსეთ ნიშნულამდე (ამ თავის პუნქტი 3.24). თუ შიდა სტანდარტი არ არის გამოყენებული, დაამატეთ 2,00 მლ შიდა სტანდარტი (ამ თავის პუნქტი 3.27.3) და შეავსეთ ნიშნულამდე ციტრატის ბუფერით (ამ თავის პუნქტი 3.24). აურიეთ საფუძვლიანად. გადადით ქრომატოგრაფიის ეტაპზე (ამ თავის პუნქტი 5.4). თუ ნიმუშის ხსნარების გამოკველვა არ ხდება იმავე დღეს, ისინი შენახული უნდა იქნეს 5oC-ზე დაბალ ტემპერატურაზე. 5.4. ქრომატოგრაფია ქრომატოგრაფიამდე, მიიყვანეთ ექსტრაქტი (ამ თავის პუნქტი 5.2) ან ჰიდროლიზატი (ამ თავის პუნქტი 5.3.4) ოთახის ტემპერატურამდე. შეანჯღრიეთ ნარევი და გაფილტრეთ საჭირო რაოდენობა 0,22 მკმ მემბრანული ფილტრის საშუალებით (ამ თავის პუნქტი 4.5). შედეგად მიღებული გამჭვირვალე ხსნარი ექვემდებარება იონგაცვლით ქრომატოგრაფიას, ამინომჟავების ანალიზატორის გამოყენებით (ამ თავის პუნქტი 4.9). ინჯექტირება შეიძლება გაკეთდეს ხელით ან ავტომატურად. მნიშვნელოვანია, რომ იმავე რაოდენობის ხსნარი ± 0,5% დაემატოს სვეტს სტანდარტებისა და ნიმუშების გამოკვლევისათვის, გარდა იმ შემთხვევისა, როდესაც გამოიყენება შიდა სტანდარტი, და რომ ნატრიუმი/ამინომჟავის თანაფარდობა სტანდარტულ და ნიმუშის ხსნარებში იყოს რაც შეიძლება პრაქტიკულად მსგავსი. ზოგადად საკალიბრო სიხშირე დამოკიდებულია ნინჰიდრინის რეაგენტისა და ანალიზური სისტემის სტაბილურობაზე. სტანდარტი ან ნიმუში განზავებულია ციტრატის ბუფერით (ამ თავის პუნქტი 3.24), რათა ამინომჟავის ნიმუშის პიკური ფართობიდან მივიღოთ სტანდარტის პიკური ფართობი 30% -200% დიაპაზონში. ამინომჟავების ქრომატოგრაფია ოდნავ შეიცვლება გამოყენებული ანალიზატორის ტიპისა და გამოყენებული ფისის შესაბამისად. არჩეულ სისტემას უნდა შეეძლოს ამინომჟავების განცალკევება ერთმანეთისაგან და ნინჰიდრინის შემცველი მასალებისგან. საოპერაციო დიაპაზონში ქრომატოგრაფიულმა სისტემამ ხაზოვანი რეაგირება უნდა მოახდინოს სვეტში დამატებული ამინომჟავების რაოდენობის ცვლილებებზე. ქრომატოგრაფიის ეტაპზე მინიმალური წერტილი: ქვემოთ აღწერილი პიკის სიმაღლის თანაფარდობა გამოიყენება, როდესაც ხდება ექვიმოლური (ტოლფასოვანი) ხსნარის (განსაზღვრული ამინომჟავები) გამოკვლევა. ეს ექვიმოლური ხსნარი უნდა შეიცავდეს თითოეული ამინომჟავის მაქსიმალური დატვირთვის სულ მცირე 30%-ს, რომლის ზუსტი გაზომვაც შესაძლებელია ამინომჟავების ანალიზატორის სისტემით (ამ თავის პუნქტი 4.9). ტრეონინ-სერინის გამოყოფისთვის მინიმალური წერტილი: ქრომატოგრამაზე ორი გადამფარავი ამინომჟავის ქვედა პიკის სიმაღლის თანაფარდობა არ უნდა აღემატებოდეს 2:10. (თუ განისაზღვრება მხოლოდ ცისტ(ე)ინი, მეთიონინი, ტრეონინი და ლიზინი, მომიჯნავე პიკების არასაკმარისი გამოყოფა უარყოფითად იმოქმედებს განსაზღვრაზე). ყველა სხვა ამინომჟავისთვის გამოყოფა უნდა იყოს 1:10 -ზე უკეთესი. სისტემამ უნდა უზრუნველყოს, რომ ლიზინი გამოყოფილია „ლიზინის არტიფაქტებისგან“ და ორნითინისგან. 6. შედეგების ინტერპრეტაცია ნიმუშის ფართობი და სტანდარტული პიკები იზომება თითოეული ამინომჟავისთვის და ამინომჟავას რაოდენობა (X) გ-ში თითო კგ ნიმუშზე გამოითვლება შემდეგნაირად:
თუ შიდა სტანდარტი გამოიყენება, გაამრავლეთ: D/C-ზე. სადაც: A = პიკის ფართობი, ჰიდროლიზატი ან ექსტრაქტი; B = პიკის ფართობი, საკალიბრო სტანდარტული ხსნარი; C = პიკის ფართობი, შიდა სტანდარტი ჰიდროლიზატში ან ექსტრაქტში; D = პიკის ფართობი, შიდა სტანდარტი, საკალიბრო სტანდარტული ხსნარი; M = განისაზღვრება ამინომჟავის მოლური წონა; c = სტანდარტის კონცენტრაცია მმოლი/მლ; m = ნიმუშის წონა (გ) (შესწორებულია თავდაპირველი წონის მისაღებად, თუ პროდუქტი გამომშრალი ან ცხიმგამოცლილია); V = ჰიდროლიზატის საერთო რაოდენობა მლ-ში (ამ თავის პუნქტი 5.3.4) ან გამოანგარიშებულია ექსტრაქტის განზავების საერთო მოცულობა მლ-ში (ამ თავის პუნქტი 6.1) ცისტინი და ცისტეინი განისაზღვრება, როგორც ცისტეინის მჟავა დაჟანგული ნიმუშის ჰიდროლიზატებში, მაგრამ გამოითვლება ცისტინად (C6H12N2O4S2, M 240,30 გ/მოლი) M-ის გამოყენებით 120,15 გ/მოლზე (= 0,5 x 240,30 გ/მოლი).
დაჟანგული ნიმუშის ჰიდროლიზატებში მეთიონინი განისაზღვრება როგორც მეთიონინის სულფონი, მაგრამ გამოითვლება როგორც მეთიონინი M მეთიონინის გამოყენებით: 149,21 გ/მოლი. დამატებული თავისუფალი მეთიონინი განისაზღვრება ექსტრაჰირების შემდეგ, როგორც მეთიონინი, გაანგარიშებისთვის გამოიყენება იმავე M. 6.1. ექსტრაქტების განზავების საერთო მოცულობა (F) თავისუფალი ამინომჟავების დასადგენად (ამ თავის პუნქტი 5.2) გამოითვლება შემდეგნაირად:
V = საბოლოო ექტრაქტის მოცულობა
7. მეთოდის შეფასება მეთოდი შემოწმებულ იქნა 1990 წელს საერთაშორისო დონეზე ჩატარებულ ურთიერთშედარებაში ოთხი განსხვავებული ცხოველის საკვების გამოყენებით (შერეული ღორის საკვები, ბროილერის კომბინირებული საკვები, პროტეინის კონცენტრატი, პრემიქსი).
შედეგები, საშუალო მნიშვნელობების და სტანდარტული გადახრის დეავიაციის აღმოფხვრის შემდეგ, მოცემულია ამ პუნქტის ცხრილებში:
წონითი მნიშნელობები გ/კგ-ში
7.1. განმეორებადობა განმეორებადობა, რომელიც გამოხატულია როგორც „ლაბორატორიული სტანდარტული გადახრის დიაპაზონში“ ზემოთ მოცემული ურთიერთშედარების მიხედვით, მოცემულია ცხრილებში:
შიდა ლაბორატორიული სტანდარტული გადახრა (Sr) გ/კგ-ში
ვარიაციის კოეფიციენტი (%) შიდა ლაბორატორიული სტანდარტული გადახრისთვის (Sr)
7.2. რეპროდუქციულობა (შედეგიანობა) ლაბორატორიათშორისი სტანდარტული გადახრის შედეგები, რომლებიც ზემოთ მოყვანილ ურთიერთშედარებშია ასახული, მოცემულია ქვემოთ მოცემულ ცხრილში:
ლაბორატორიათშორისი სტანდარტული გადახრა (SR) გ/კგ
ვარიაციის (%) ლაბორატორიათშორისი სტანდარტული გადახრისთვის (SR)
8. ეტალონური (რეფერენს) მასალის გამოყენება მეთოდის სწორი გამოყენება უნდა გადამოწმდეს (ვერიფიკაცია) სერტიფიცირებული რეფერენსმასალების განმეორებითი გაზომვებით, ასეთი არსებობის შემთხვევაში. დაკალიბრება რეკომენდებულია განხორციელდეს ამინომჟავების საკალიბრო სერტიფიცირებული ხსნარით. 9. დაკვირვება 9.1. ამინომჟავების ანალიზატორებს შორის განსხვავებების გამო, სტანდარტული ამინომჟავების (იხ. ამ თავის პუნქტი 3.27.4 და 3.27.5) და ჰიდროლიზატის (იხ. ამ თავის პუნქტი 5.3.4) საკალიბრო ხსნარების საბოლოო კონცენტრაცია მიღებული უნდა იქნეს როგორც სახელმძღვანელო პრინციპი. აპარატის/დანადგარის ხაზოვანი რეაგირების დიაპაზონი უნდა შემოწმდეს ყველა ამინომჟავასთვის. სტანდარტული ხსნარი ზავდება ციტრატის ბუფერით, რათა მიღებულ იქნეს პიკური ფართობი შუა დიაპაზონში. 9.2. როდესაც ჰიდროლიზატების გასაანალიზებლად გამოიყენება მაღალი ხარისხის თხევადი ქრომატოგრაფიული მოწყობილობა, ექსპერიმენტული პირობები უნდა იყოს ოპტიმიზირებული მწარმოებლის რეკომენდაციების შესაბამისად. 9.3. ამ მეთოდის გამოყენებამ 1%-ზე მეტი ქლორიდის შემცველი ცხოველის საკვებზე (კონცენტრატი, ცხოველის მინერალური საკვები, დამატებითი საკვები) შეიძლება გამოიწვიოს მეთიონინის არასწორი შეფასება, და სპეციალური დამუშავების ჩატარება გახდება საჭირო.
თავი VII ტრიფტოფანის განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა ცხოველის საკვებში განვსაზღვროთ ტრიფტოფანის საერთო შემცველობა და თავისუფალი ტრიფტოფანი. იგი არ განასხვავებს D- და L- ფორმებს. 2. გამოყენების პრინციპი ტრიფტოფანის საერთო შემცველობის განსაზღვრისთვის, ნიმუში ჰიდროლიზდება ტუტე პირობებში გაჯერებული ბარიუმის ჰიდროქსიდის ხსნარით და თბება 110oC ტემპერატურაზე 20 საათის განმავლობაში. ჰიდროლიზის შემდეგ ემატება შიდა სტანდარტი. თავისუფალი ტრიფტოფანის განსაზღვრისთვის, ხდება ნიმუშის ექსტრაჰირება მსუბუქი მჟავას გარემოში, შიდა სტანდარტთან ერთად. ჰიდროლიზატში ან ექსტრაქტში ტრიფტოფანი და შიდა სტანდარტი განისაზღვრება HPLC-ით, ფლუორესცენტის გამოვლენით. 3. რეაგენტები 3.1. გამოყენებული უნდა იქნეს ორმაგად გამოხდილი წყალი ან ეკვივალენტური ხარისხის წყალი (გამტარობა <10 მკ S/სმ). 3.2. სტანდარტული ნივთიერება: ტრიფტოფანის (სისუფთავე/შემცველობა ≥ 99%) გაშრობა წარმოებს ვაკუუმში ფოსფორის პენტოქსიდის გამოყენებით. 3.3. შიდა სტანდარტული ნივთიერება: α-მეთილ-ტრიფტოფანის (სისუფთავე/შემცველობა ≥ 99%) გაშრობა წარმოებს ვაკუუმში ფოსფორის პენტოქსიდის გამოყენებით. 3.4. ბარიუმის ჰიდროქსიდის ოქტა-ჰიდრატი (ყურადღება მიაქცეთ, რომ Ba (OH)2 .8 H2O არ დაექვემდებაროს ჰაერის ჭარბ ზემოქმედებას, რათა თავიდან იქნეს აცილებული BaCO3-ის წარმოქმნა, რამაც შეიძლება ხელი შეუშალოს განსაზღვრას) (იხ. დაკვირვება ამ თავის პუნქტი 9.3). 3.5. ნატრიუმის ჰიდროქსიდი. 3.6. ორთო-ფოსფორის მჟავა, w (w/w) = 85%. 3.7. მარილმჟავა, ρ 20 1,19 გ/მლ. 3.8. მეთანოლი, HPLC ხარისხის ეკვივალენტი. 3.9. პეტროლეუმის ეთერი, დუღილის დიაპაზონი 40-60oC. 3.10. ნატრიუმის ჰიდროქსიდის ხსნარი, c = 1 მოლი/ლ: გახსენით 40.0 გ NaOH (ამ თავის პუნქტი 3.5) წყალში და შეავსეთ 1 ლიტრამდე წყლით (ამ თავის პუნქტი 3.1). 3.11. მარილმჟავა, c = 6 მოლი/ლ: აიღეთ 492 მლ HCl (ამ თავის პუნქტი 3.7) და წყლით შეავსეთ 1 ლიტრამდე. 3.12. მარილმჟავა, c = 1 მოლი/ლ: აიღეთ 82 მლ HCl (ამ თავის პუნქტი 3.7) და შეავსეთ 1 ლიტრამდე, წყლით. 3.13. მარილმჟავა, c = 0,1 მოლი/ლ: აიღეთ 8,2 მლ HCl (ამ თავის პუნქტი 3.7) და შეავსეთ 1 ლიტრამდე წყლით. 3.14. ორთო-ფოსფორის მჟავა, c = 0,5 მოლ/ლ: აიღეთ 34 მლ ორთო-ფოსფორის მჟავა (ამ თავის პუნქტი 3.6) და შეავსეთ 1 ლიტრამდე წყლით (ამ თავის პუნქტი 3.1). 3.15. ტრიფტოფანის კონცენტრირებული ხსნარი (3.2), c = 2,50 მკმოლ/მლ: 500 მლ მოცულობით კოლბაში გახსენით 0,2553 გ ტრიფტოფანი (ამ თავის პუნქტი 3.2) მარილმჟავაში (ამ თავის პუნქტი 3.13) და შეავსეთ ნიშნულამდე მარილმჟავათი (ამ თავის პუნქტი 3.13). შეინახეთ -18oC ტემპერატურაზე არაუმეტეს 4 კვირის განმავლობაში. 3.16. კონცენტრირებული შიდა სტანდარტული ხსნარი, c = 2,50 მკმოლ/მლ: 500 მლ მოცულობის კოლბაში გახსენით 0,2728 გ α-მეთილ-ტრიფტოფანი (ამ თავის პუნქტი 3.3) მარილმჟავაში (ამ თავის პუნქტი 3.13) და შეავსეთ ნიშნულამდე მარილმჟავათი (ამ თავის პუნქტი 3.13). შეინახეთ - 18oC ტემპერატურაზე მაქსიმუმ 4 კვირის განმავლობაში. 3.17. ტრიფტოფანის და შიდა სტანდარტის საკალიბრო სტანდარტული ხსნარი: აიღეთ 2,00 მლ ტრიფტოფანის კონცენტრირებული ხსნარი (ამ თავის პუნქტი 3.15), და 2,00 მლ კონცენტრირებული შიდა სტანდარტის (α-მეთილ-ტრიფტოფანი) ხსნარი (ამ თავის პუნქტი 3.16). განაზავეთ წყლით (ამ თავის პუნქტი 3.1) და მეთანოლით (ამ თავის პუნქტი 3.8) დაახლოებით იმავე მოცულობისა და დაახლოებით იმავე მეთანოლის კონცენტრაციის (10% -30%) მისაღწევად, როგორც საბოლოო ჰიდროლიზატი. გამოყენებამდე ეს ხსნარი უნდა იყოს ახლი მომზადებული. მომზადების დროს დაიცავით მზის პირდაპირი სხივებისგან. 3.18. ძმარმჟავა. 3.19. 1,1,1-ტრიქლორო-2-მეთილ-2-პროპანოლი. 3.20. ეთანოლამინი w (w/w)> 98%. 3.21. 1 გ 1,1,1-ტრიქლორო-2-მეთილ-2-პროპანოლის (ამ თავის პუნქტი 3.19) ხსნარი 100 მლ მეთანოლში (ამ თავის პუნქტი 3.8). 3.22. მობილური ფაზა HPLC-სთვის: 3,00 გ ძმარმჟავა (ამ თავის პუნქტი 3.18) + 900 მლ წყალი (ამ თავის პუნქტი 3.1) + 50,0 მლ ხსნარი (ამ თავის პუნქტი 3.21) 1,1,1-ტრიქლორ-2-მეთილ-2-პროპანოლის (ამ თავის პუნქტი 3.19) მეთანოლში (ამ თავის პუნქტი 3.8) (1 გ/100 მლ). pH-ის მნიშვნელობა დააკორექტირეთ 5,00-ზე ეთანოლამინის გამოყენებით (ამ თავის პუნქტი 3.20). შეავსეთ 1000 მლ-მდე, წყლით (ამ თავის პუნქტი 3.1). 4. აპარატურა 4.1. HPLC მოწყობილობა სპექტროფლუორომეტრიული დეტექტორით. 4.2. თხევადი ქრომატოგრაფიული სვეტი, 125 მმ x 4 მმ, C 18, 3 მკმ შეფუთვა, ან ეკვივალენტი. 4.3. pH მეტრი. 4.4. პოლიპროპილენის კოლბა, ტევადობა 125 მლ, ფართო ყელითა და ხრახნიანი თავსახურით. 4.5. მემბრანული ფილტრი, 0,45 მკმ. 4.6. ავტოკლავი, 110 (± 2)oC, 1,4 (± 0,1) ბარი. 4.7. მექანიკური სადღვები ან მაგნიტური სათქვეფელა. 4.8. ვორტექს (შემრევი) მიქსერი. 5. პროცედურა 5.1. ნიმუშების მომზადება ნიმუში ქუცმაცდება, რათა გატარდეს 0.5 მმ-იანი ხვრელების მქონე საცერში. მაღალი ტენიანობის შემცველი ნიმუშები უნდა გაშრეს, ან ჰაერის ნაკადით არაუმეტეს 50 oC ტემპერატურაზე, ან უნდა მოხდეს მათი ლიოფიზირება დაფქვის წინ. მაღალი რაოდენობის ცხიმშემცველი ნიმუშების ექსტრაჰირება ხდება პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.9) დაქუცმაცებამდე. 5.2. თავისუფალი ტრიფტოფანის (ექსტრაქტის) განსაზღვრა აწონეთ 1 მგ სიზუსტით, შესაბამისი რაოდენობით (1-5 გ) მომზადებული ნიმუში (ამ თავის პუნქტი 5.1), კონუსურ კოლბაში. დაამატეთ 100,0 მლ მარილმჟავა (ამ თავის პუნქტი 3.13) და 5,00 მლ კონცენტრირებული შიდა სტანდარტული ხსნარი (ამ თავის პუნქტი 3.16). ანჯღრიეთ ან ათქვიფეთ 60 წუთის განმავლობაში, მექანიკური სადღვების ან მაგნიტური სათქვეფელას გამოყენებით (ამ თავის პუნქტი 4.8). მიეცით ნალექს დაწმენდის (დალექვის) საშუალება და 10,0 მლ ნალექზედა ხსნარის შრე (სუპერნატანტი) პიპეტით გადაიტანეთ მენზურაში. დაამატეთ 5 მლ ორთო-ფოსფორის მჟავა (ამ თავის პუნქტი 3.14). pH დააკორექტირეთ 3-ზე ნატრიუმის ჰიდროქსიდის გამოყენებით (ამ თავის პუნქტი 3.10). დაამატეთ საკმარისი რაოდენობის მეთანოლი (ამ თავის პუნქტი 3.8), რათა საბოლოო მოცულობაში მივიღით 10% და 30% მეთანოლის კონცენტრაცია. გადაიტანეთ შესაბამისი მოცულობის დანაყოფებიან (გრადირებულ) კოლბაში და განაზავეთ წყლით ქრომატოგრაფიისთვის საჭირო მოცულობამდე (დაახლოებით იმავე მოცულობა, როგორც საკალიბრო სტანდარტული ხსნარისა (ამ თავის პუნქტი 3.17). HPLC-ის სვეტის ინჯექტირებამდე გაფილტრეთ რამდენიმე მლ ხსნარი 0,45 მკმ გარსის ფილტრის საშუალებით (ამ თავის პუნქტი 4.5). გადადით ქრომატოგრაფიის ეტაპზე ამ თავის 5.4 პუნქტის შესაბამისად. დაიცავით სტანდარტული ხსნარი და ექსტრაქტები მზის პირდაპირი სხივებისგან. თუ შეუძლებელია ექსტრაქტების გამოკვლევა იმავე დღეს, შესაძლებელია ექსტრაქტების შენახვა 5 oC ტემპერატურაზე არაუმეტეს 3 დღისა. 5.3. ტრიფტოფანის საერთო რაოდენობის (ჰიდროლიზატის) განსაზღვრა პოლიპროპილენის კოლბაში (ამ თავის პუნქტი 4.4) აწონეთ 0,2 მგ სიზუსტით 0,1-დან 1 გ-მდე მომზადებული ნიმუში (ამ თავის პუნქტი 5.1). შეწონილი ნიმუშის პორციას უნდა ჰქონდეს დაახლოებით 10 მგ-ის აზოტის შემცველობა. დაამატეთ 8,4 გ ბარიუმის ჰიდროქსიდის ოქტა-ჰიდრატი (ამ თავის პუნქტი 3.4) და 10 მლ წყალი. აურიეთ ვორტექს (შემრევი) მიქსერით (ამ თავის პუნქტი 4.8) ან მაგნიტუროს შემრევით (ამ თავის პუნქტი 4.7). ტეფლონით დაფარული მაგნიტი დატოვეთ ნარევში. გარეცხეთ ჭურჭლის კედლები 4 მლ წყლით. გაუკეთეთ ხრახნიანი თავსახური და კოლბა მოშვებულად დახურეთ (არაჰერმეტულად). გადაიტანეთ მდუღარეწყლიან ავტოკლავში (ამ თავის პუნქტი 4.6) და ამყოფეთ ორთქლის ზემოქმედების ქვეშ 30-60 წუთის განმავლობაში. დახურეთ ავტოკლავი და მოახდინეთ ავტოკლავირება (სტერილიზება) 110 (± 2) o C ტემპერატურაზე 20 საათის განმავლობაში. ავტოკლავის გახსნამდე დააყენეთ ტემპერატურა 100°C-ის ქვემოთ. იმისათვის, რომ თავიდან იქნეს აცილებული Ba(OH)2 · 8 H2O-ის კრისტალიზაცია, თბილ ნარევს დაუმატეთ ოთახის ტემპერატურის მქონე 30 მლ წყალი. შეანჯღრიეთ ან აურიეთ ნაზად. დაამატეთ 2,00 მლ კონცენტრირებული შიდა სტანდარტის (α-მეთილ-ტრიფტოფანი) ხსნარი (ამ თავის პუნქტი 3.16). გააგრილეთ ჭურჭელი წყლის/ყინულის აბაზანაში 15 წუთის განმავლობაში. შემდეგ დაამატეთ 5 მლ ორთო-ფოსფორის მჟავა (ამ თავის პუნქტი 3.14). შეინახეთ ჭურჭელი გამაგრილებელ აბაზანაში და გაანეიტრალეთ HCl საშუალებით (ამ თავის პუნქტი 3.11), თან ურიეთ და pH დააკორექტირეთ 3,0-მდე HCl-ის გამოყენებით (ამ თავის პუნქტი 3.12). დაამატეთ საკმარისი რაოდენობის მეთანოლი, რომ საბოლოო მოცულობაში მივიღით 10% და 30% მეთანოლის კონცენტრაცია. გადაიტანეთ შესაბამისი მოცულობის დანაყოფებიან (გრადირებულ) კოლბაში და განაზავეთ წყლით ქრომატოგრაფიისთვის საჭირო მოცულობამდე (მაგალითად 100 მლ). მეთანოლის დამატებამ არ უნდა გამოიწვიოს ნალექის წარმოშობა. HPLC-ის სვეტის ინჯექტირებამდე, 0,45 მკმ გარსის ფილტრის საშუალებით (ამ თავის პუნქტი 4.5) გაფილტრეთ რამდენიმე მლ ხსნარი. გადადით ქრომატოგრაფიის ეტაპზე ამ თავის 5.4 პუნქტის შესაბამისად. დაიცავით სტანდარტული ხსნარი და ჰიდროლიზატები მზის პირდაპირი სხივებისგან. თუ ჰიდროლიზატების გამოკვლევა შეუძლებელია იმავე დღეს, მათი შენახვა შესაძლებელია 5 oC ტემპერატურაზე არაუმეტეს მაქსიმუმ 3 დღის განმავლობაში. 5.4. HPLC-ის განსაზღვრა სახელმძღვანელოდ შემოთავაზებულია იზოკრატიული ელუციის შემდეგი პირობები; შეიძლება გამოყენებულ იქნეს სხვა პირობები, იმ პირობით, თუ მათ მივყავართ ეკვივალენტურ შედეგებამდე (იხილეთ აგრეთვე ამ თავის პუნქტები 9.1 და 9.2):
6. შედეგების ინტერპრეტაცია ტრიფტოფანის (X) რაოდენობა, გ-ში 100 გ ნიმუშზე, გამოითვლება შემდეგნაირად:
სადაც: A = შიდა სტანდარტის პიკური ფართობი, საკალიბრო სტანდარტული ხსნარი (ამ თავის პუნქტი 3.17); B = ტრიფტოფანის პიკური ფართობი, ექსტრაქტი (ამ თავის პუნქტი 5.2) ან ჰიდროლიზატი (ამ თავის პუნქტი 5.3); V1 = მოცულობა მლ (2 მლ)-ში კონცენტრირებული ტრიფტოფანის ხსნარის (ამ თავის პუნქტი 3.15), რომელიც დაემატა საკალიბრო ხსნარს (ამ თავის პუნქტი 3.17); c = კონცენტრაცია მკ მოლ/ მლ (= 2,50)-ში კონცენტრირებული ტრიფტოფანის ხსნარისა (ამ თავის პუნქტი 3.15), რომელიც დაემატა საკალიბრო ხსნარს (ამ თავის პუნქტი 3.17); V2 = მოცულობა მლ-ში კონცენტრირებული შიდა სტანდარტული ხსნარისა (3.16), რომელიც დაემატა ექსტრაჰირებისას (ამ თავის პუნქტი 5.2) (= 5,00 მლ) ან ჰიდროლიზატს (ამ თავის პუნქტი 5.3) (= 2,00 მლ); C = შიდა სტანდარტის პიკური ფართობი, ექსტრაქტი (ამ თავის პუნქტი 5.2) ან ჰიდროლიზატი (ამ თავის პუნქტი 5.3); D = ტრიფტოფანის პიკური ფართობი, საკალიბრო სტანდარტული ხსნარი (ამ თავის პუნქტი 3.17); V3 = მოცულობა მლ-ში (= 2,00 მლ) კონცენტრირებული შიდა სტანდარტული ხსნარისა (ამ თავის პუნქტი 3.16), რომელიც დაემატა საკალიბრო სტანდარტულ ხსნარს (ამ თავის პუნქტი 3.17). m = ნიმუშის წონა გ-ში (კორექტირებული თავდაპირველ წონაზე, თუ გამომშრალია და/ან ცხიმგამოცლილია) M = ტრიფტოფანის მოლური წონა (= 204,23 გ/მოლი)
7. განმეორებადობა ერთი და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 10%-ს ყველაზე მაღალ შედეგთან შედარებისას. 8. ერთობლივი კვლევის შედეგები ჰიდროლიზის მეთოდის დასადასტურებლად (სერტიფიცირებისათვის) მოეწყო კიდევ ერთი ევროკომისიის ერთობლივი კვლევა (მე-4 ურთიერთშედარება), რომლის დროსაც გამოკვლეულ იქნა სამი ნიმუში 12-მდე ლაბორატორიაში. თითოეულ ნიმუშზე ჩატარდა რეპლიკაციური (განმეორებითი) (5) გამოკვლევა. შედეგები მოცემულია ქვემოთ, ცხრილში:
L = შედეგების წარმდგენი ლაბორატორიების რაოდენობა; n = ცალკეული შედეგების რაოდენობა, რომლებიც არის შენაჩუნებული გადახრების; აღმოფხვრის შემდეგ (იდენტიფიცირებული Cochran, Dixon გადახრის ტესტით მიხედვით); Sr = განმეორებადობის სტანდარტული გადახრა; S R = რეპროდუქციულობის (შედეგიანობის)სტანდარტული გადახრა; r = განმეორებადობა; R = რეპროდუქციულობა (შედეგიანობა); CV r = განმეორებადობის ვარიაციის კოეფიციენტი, %; CV R = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი, %;
თავისუფალი ტრიფტოფანის ექსტრაციის დასადასტურებლად (სერტიფიცირებისათვის) მოეწყო კიდევ ერთი ევროკომისიის ერთობლივი კვლევა (მე-3 ურთიერთშედარება), რომლის დროსაც გამოკვლეულ იქნა სამი ნიმუში 13-მდე ლაბორატორიაში. თითოეულ ნიმუშზე ჩატარდა რეპლიკაციური (განმეორებითი) (5) გამოკვლევა. შედეგები მოცემულია ქვემოთ, ცხრილში:
L = შედეგების წარმდგენი ლაბორატორიების რაოდენობა; n = ცალკეული შედეგების რაოდენობა, რომლებშიც არის შენარჩუნებული გადახრების აღმოფხვრის შემდეგ (იდენტიფიცირებული Cochran, Dixon გადახრის ტესტით მიხედვით); Sr = განმეორებადობის სტანდარტული გადახრა; S R = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; r = განმეორებადობა; R = რეპროდუქციულობა (შედეგიანობა); CV r = განმეორებადობის ვარიაციის კოეფიციენტი, %; CV R = რეპროდუქციულობის ვარიაციის კოეფიციენტი, %;
ჰიდროლიზის მეთოდის დასადასტურებლად მოეწყო კიდევ ერთი ევროკომისიის ერთობლივი კვლევა, რომლის დროსაც გამოკვლეულ იქნა 4 ნიმუში 7-მდე ლაბორატორიაში. თითოეულ ნიმუშზე ჩატარდა რეპლიკაციური (განმეორებითი) (5) გამოკვლევა ტრიფტოფანის სერტიფიცირების მიზნით. შედეგები მოცემულია ქვემოთ, ცხრილში:
L = შედეგების წარმდგენი ლაბორატორიების რაოდენობა; n = ცალკეული შედეგების რაოდენობა, რომლებშიც არის შენაჩუნებული გადახრების აღმოფხვრის შემდეგ (იდენტიფიცირებული Cochran, Dixon გადახრის ტესტით მიხედვით); Sr = განმეორებადობის სტანდარტული გადახრა; SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; r = განმეორებადობა; R = რეპროდუქციულობა (შედეგიანობა); CV r = განმეორებადობის ვარიაციის კოეფიციენტი, %; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი, %;
9. დაკვირვება 9.1. შემდგომმა სპეციალურმა ქრომატოგრაფიულმა პირობებმა შეიძლება მოგვცეს უკეთესი გაყოფა ტრიფტოფანსა და α-მეთილ-ტრიფტოფანს შორის.
იზოკრატიული ელუცია, რასაც თან სდევს გრადიენტური სვეტის გაწმენდა:
9.2. HPLC ტიპისა და გამოყენებული სვეტის შესაფუთი მასალის მიხედვით, ქრომატოგრაფია მოგვცემს განსხვავებას. არჩეულმა სისტემამ უნდა უზრუნველყოს ტრიფტოფანისა და შიდა სტანდარტის საბაზისო გაყოფა. უფრო მეტიც, მნიშვნელოვანია, რომ დეგრადაციის პროდუქტები კარგად იყოს განცალკავებული ტრიფტოფანისა და შიდა სტანდარტისგან. ჰიდროლიზატები შიდა სტანდარტის გარეშე უნდა იქნეს გამოყენებული, შიდა სტანდარტში საბაზისო ხაზის მინარევების არსებობაზე შემოწმების მიზნით. მნიშვნელოვანია, რომ გამოკვლევის დრო იყოს საკმარისად ხანგრძლივი ყველა დეგრადაციის პროდუქტის გასათავისუფლებლად, წინააღმდეგ შემთხვევაში, დაგვიანებულმა ელუციის პიკებმა შეიძლება ხელი შეუშალოს შემდგომ ქრომატოგრაფიულ გამოკვლევებს. მოქმედების დიაპაზონში, ქრომატოგრაფიული სისტემა უნდა იძლეოდეს ხაზოვან რეაქციას. წრფივი პასუხი უნდა იზომებოდეს შიდა სტანდარტის მუდმივი (ნორმალური) კონცენტრაციით და ტრიფტოფანის ცვალებადი კონცენტრაციით. მნიშვნელოვანია, რომ ტრიფტოფანისა და შიდა სტანდარტის მწვერვალების ზომა იყოს HPLC/ფლუორესენციის სისტემის წრფივ დიაპაზონში. თუ ტრიფტოფანი და/ან შიდა სტანდარტული პიკი(ები) ძალიან მცირე ან ძალიან მაღალია, გამოკვლევა უნდა განმეორდეს სხვა ნიმუშის ზომით და/ან შეცვლილი საბოლოო მოცულობით. 9.3. ბარიუმის ჰიდროქსიდი დროთა განმავლობაში ბარიუმის ჰიდროქსიდის გახსნა უფრო ძნელდება. ეს HPLC-ის განსაზღვრისთვის იწვევს ისეთი ბუნდოვანი ხსნარის მიღებას, რამაც შეიძლება მოგვცეს დაბალი შედეგები ტრიფტოფანისთვის. თავი VIII ნედლი ზეთებისა და ცხიმების განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი განკუთვნილია ცხოველის საკვებში ნედლი ზეთებისა და ცხიმების განსაზღვრისთვის. იგი არ მოიცავს ზეთოვანი კულტურების თესლისა და კაკლოვანი ნაყოფის გამოკვლევას. ქვემოთ აღწერილი ორი პროცედურის გამოყენება დამოკიდებულია ცხოველის საკვების ბუნებასა და შემადგენლობაზე, და გამოკვლევის ჩატარების მიზეზზე. 1.1. პროცედურა A – პირდაპირ ექსტრაჰირებადი ნედლი ზეთები და ცხიმები ეს მეთოდი გამოიყენება მცენარეული წარმოშობის ცხოველის საკვების მასალებზე, გარდა იმ პროცედურისა, რაც აღწერილია B პროცედურაში. 1.2. პროცედურა B – ნედლი ზეთებისა და ცხიმების საერთო რაოდენობა ეს მეთოდი გამოიყენება ცხოველური წარმოშობის საკვების მასალებზე და ცხოველის ყველა კომბინირებული საკვებისთვის. იგი გამოყენებული უნდა იქნეს ყველა მასალისთვის, რომლიდანაც ზეთებისა და ცხიმების სრულად ექსტრაჰირება შეუძლებელია წინასწარი ჰიდროლიზის გარეშე (მაგ., წებოვანა, საფუარი, კარტოფილის ცილები და პროდუქტები, რომლებიც ექვემდებარება ექსტრუზიას, აქერცვლას და გათბობას). 1.3. შედეგების ინტერპრეტაცია ყველა შემთხვევაში, როდესაც B პროცედურის გამოყენებით მიიღება უფრო მაღალი შედეგი, ვიდრე A პროცედურის გამოყენებით, B პროცედურის შედეგად მიღებული შედეგი მიღებული უნდა იქნეს, როგორც რეალური მნიშვნელობა.
2. გამოყენების პრინციპი 2.1. პროცედურა A ნიმუშის ექსტრაჰირება ხდება პეტროლეუმის ეთერით. გამხსნელს აცალკავებენ გამოხდის გზით და ხდება ნაშთის (ნარჩენების) გაშრობა და აწონვა.
2.2. პროცედურა B ნიმუშების მუშავდება გაცხელების ქვეშ მარილმჟავათი. ნარევი გრილდება და იფილტრება. ნაშთი (ნარჩენები) ირეცხება და შრება და ხდება მათი წარდგენა განსაზღვრისთვის A პროცედურის მიხედვით.
3. რეაგენტები 3.1. პეტროლეუმის ეთერი, დუღილის დიაპაზონი: 40-დან 60oC. ბრომის მნიშვნელობა უნდა იყოს 1-ზე ნაკლები, ხოლო აორთქლებისას ნაშთი (ნარჩენები) 2 მგ/100 მლ-ზე ნაკლები. 3.2. ნატრიუმის სულფატი, უწყლო. 3.3. მარილმჟავა, c = 3 მოლი/ლ. 3.4. ფილტრაციის დამხმარე საშუალება, მაგ. Kieselguhr, Hyflo-supercel.
4. აპარატურა 4.1. ექსტრაჰირების აპარატი/დანადგარი. თუ მას აქვს სიფონი (Soxhlet აპარატი), რეფლუქსის სიჩქარე უნდა იყოს ისეთი, რომ საათში დაახლოებით 10 ციკლი წარმოიქმნას; თუ არ არის სიფონირებული ტიპი, რეფლუქსის სიჩქარე უნდა იყოს დაახლოებით 10 მლ წუთში. 4.2. ექსტრაჰირების კარტუში, რომელიც პეტროლეუმის ეთერში თავისუფალია ხსნადი მასალისაგან და გააჩნია ამ თავის 4.1 პუნქტის მოთხოვნებების შესაბამისი ფორიანობა. 4.3. საშრობი ღუმელი, ან ვაკუუმური ღუმელი დარეგულირებული 75 ± 3o C ტემპერატურაზე, ან ჰაერის ღუმელი დარეგულირებული 100 ± 3o C ტემპერატურაზე.
5. პროცედურა 5.1. პროცედურა A (იხ. ამ თავის პუნქტი 8.1)
აწონეთ 5 გ ნიმუში 1 მგ-მდე სიზუსტით, გადაიტანეთ ექსტრაქციის კარტუშში (ამ თავის პუნქტი 4.2) და გადააფარეთ ცხიმისგან თავისუფალი ბამბის დისკები.
მოათავსეთ კარტუში ექსტრაჰირების აპარატში (ამ თავის პუნქტი 4.1) და პეტროლეუმის ეთერით მოახდინეთ ექსტრაჰირება ექვსი საათის განმავლობაში (ამ თავის პუნქტი 3.1). შეაგროვეთ პეტროლეუმის ეთერის ექსტრაქტი მშრალ, აწონილ კოლბაში, რომელიც შეიცავს პემზის ქვების ფრაგმენტებს (5).
გამოხდის მეთოდით განაცალკევეთ გამხსნელი. კოლბის საათნახევარის განმავლობაში საშრობ ღუმელში დატოვებით გააშრეთ ნაშთი (ნარჩენი) (ამ თავის პუნქტი 4.3). დატოვეთ გასაგრილებელ აპარატში და აწონეთ. კვლავ გააშრეთ 30 წუთის განმავლობაში, რომ ზეთებისა და ცხიმების წონა უცვლელი დარჩეს (წონის დაკარგვა ორ თანმიმდევრულ აწონვებს შორის უნდა იყოს 1 მგ-ზე ნაკლები ან ტოლი).
5.2. პროცედურა B აწონეთ 2,5 გ ნიმუში 1 მგ-მდე სიზუსტით (იხ. ამ თავის პუნქტი 8.2), მოათავსეთ 400 მლ მენზურაში ან 300 მლ კონუსურ კოლბაში და დაამატეთ 100 მლ მარილმჟავა (3.3) და პემზის ქვის ფრაგმენტები. დაფარეთ მენზურას საათის მინა ან მოარგეთ კონუსური კოლბა (ერლენმაიერის კოლბა) უკუმაცივრით (რეფლუქსის კონდესატორით). ნარევი მიყვანილი უნდა იქნეს ნელ დუღილამდე ცეცხლის დაბალ პარზე ან ცხელ ფირფიტაზე და ასეთ მდგომარეობაში ჩერდება ერთი საათის განმავლობაში. არ დაუშვათ, რომ პროდუქტი მიეკრას კონტეინერის გვერდებზე.
გააგრილეთ და დაამატეთ ფილტრაციის საკმარისი რაოდენობა (ამ თავის პუნქტი 3.4), რათა ფილტრაციის დროს თავიდან იქნეს აცილებული ზეთისა და ცხიმის დანაკარგი. გაფილტრეთ დატენიანებული, ცხიმგამოცლილი, ორმაგი ფილტრაციის ქაღალდით. ჩამორეცხეთ ნაშთი (ნარჩენები) ცივ წყალში, სანამ არ მიიღებთ ნეიტრალურ ფილტრატს. შეამოწმეთ, რომ ფილტრატი არ შეიცავს ზეთს ან ცხიმებს. მათი არსებობა მიუთითებს იმაზე, რომ ნიმუში უნდა იქნეს ექსტრაქტირებული პეტროლეუმის ეთერით, A პროცედურის გამოყენებით, ჰიდროლიზამდე.
ნარჩენების შემცველი ორმაგი ფილტრაციის ქაღალდი მოათავსეთ საათის მინაზე და გააშრეთ საათნახევარის განმავლობაში საჰაერო ღუმელში (ამ თავის პუნქტი 4.3) 100±3o C ტემპერატურაზე.
ორმაგი ფილტრაციის ქაღალდი, რომელიც შეიცავს მშრალ ნაშთს (ნარჩენებს), მოათავსეთ ექსტრაციის კარტუშში (ამ თავის პუნქტი 4.2) და გადააფარეთ ცხიმისგან თავისუფალი ბამბის დისკი. მოათავსეთ კარტუში ექსტრაქციის ხელსაწყოში (ამ თავის პუნქტი 4.1) და იმოქმედეთ, ისე როგორც ეს მითითებულია ამ თავის 5.1 პუნქტის მეორე და მესამე ქვეპუნქტებში.
6. შედეგების გამოხატვა ნაშთის (ნარჩენების) წონა გამოხატეთ, როგორც ნიმუშის პროცენტული მაჩვენებელი.
7. განმეორებადობა ერთი და იმავე მკვლევარის მიერ ერთსა და იმავე ნიმუშზე განხორციელებულ ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – 0,2%-ს, აბსოლუტურ მნიშვნელობაში, 5%-ზე ნაკლები ნედლი ზეთებისა და ცხიმების შემცველობისთვის, – 4,0%-ს შედარებით მაღალ შედეგებთან მიმართებაში 5%-დან 10%-მდე შემცველობისთვის, – 0,4%-ს, აბსოლუტური მნიშვნელობაში, 10% -ზე მეტი შემცველობისთვის.
8. დაკვირვება 8.1. ზეთისა და ცხიმების მაღალი შემცველობის მქონე პროდუქტებისთვის, რომელთა დაქუცმაცება ძნელია ან გამოუსადეგარია ჰომოგენიზებული შემცირებული ნიმუშის მისაღებად, იმოქმედეთ შემდეგნაირად:
აწონეთ 20 გ ნიმუში 1 მგ-მდე სიზუსტით და აურიეთ 10 გ ან მეტი უწყლო ნატრიუმის სულფატთან (ამ თავის პუნქტი 3.2). მოახდინეთ ექსტრაჰირება პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.1), როგორც ეს მითითებულია ამ თავის 5.1 პუნქტში. მიღებული ექსტრაქტი შეავსეთ 500 მლ-მდე პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.1) და აურიეთ. აიღეთ 50 მლ ხსნარი და მოათავსეთ პატარა, მშრალ, შეწონილ კოლბაში, რომელიც შეიცავს პემზის ქვის ფრაგმენტებს. გამხსნელი გამოაცალკევეთ გამოხდის მეთოდით, გააშრეთ და გააგრძელეთ, როგორც ეს მითითებულია ამ თავის 5.1 პუნქტის ბოლო ქვეპუნქტში.
გამოყავით გამხსნელი ექსტრაქტის ნაშთისგან (ნარჩენებისგან), რომელიც დარჩა კარტუშში, დააქუცმაცეთ ნარჩენი 1 მმ-მდე, დააბრუნეთ იგი ექსტრაქციის კარტუშში (არ დაამატოთ ნატრიუმის სულფატი) და გააგრძელეთ როგორც ეს მითითებულია ამ თავის 5.1 პუნქტის მეორე და მესამე ქვეპუნქტში.
ზეთებისა და ცხიმების შემცველობა გამოთვალეთ, როგორც ნიმუშის პროცენტული მაჩვენებელი შემდეგი ფორმულის გამოყენებით:
სადაც: m1 = ნაშთის (ნარჩენების) წონა გრამებში პირველი ექსტრაჰირების შემდეგ (ექსტრაქტის ფრაქციული ნაწილი); m2 = ნაშთის (ნარჩენების) წონა გრამებში მეორე ექსტრაჰირების შემდეგ. 8.2. ზეთებისა და ცხიმების დაბალი შემცველობის პროდუქტებში გამოსაკვლევი ნიმუში შეიძლება გაიზარდოს 5 გ-მდე. 8.3. შინაური ბინადარი ცხოველების საკვებს, რომელიც შეიცავს წყლის დიდ შემცველობას, შეიძლება დასჭირდეს უწყლო ნატრიუმის სულფატის შერევა ჰიდროლიზისა და ექსტრაქტირების წინ, B პროცედურის შესაბამისად. 8.4. ამ თავის 5.2 პუნქტში, ფილტრაციის შემდეგ ნაშთის (ნარჩენების) გასარეცხად, შეიძლება უფრო ეფექტური აღმოჩნდეს ცივი წყლის მაგივრად ცხელი წყლის გამოყენება. 8.5. გაშრობის დრო 1,5 სთ შეიძლება საჭირო გახდეს, რომ გაგრძელდეს ზოგიერთ ცხოველის საკვებზე. თავიდან უნდა იქნეს აცილებული გადაჭარბებული გამოშრობა, რადგან ამან შეიძლება მიგვიყვანოს დაბალ შედეგებამდე. ასევე შესაძლებელია მიკროტალღური ღუმელის გამოყენებაც. 8.6. წინასწარი ექსტრაქტირება A პროცედურით, ჰიდროლიზის ჩატარებამდე და განმეორებითი ექსტრაქტირება B პროცედურით, რეკომენდებულია, თუ ნედლი ზეთის/ცხიმის შემცველობა 15% -ზე მეტია. გარკვეულწილად ეს დამოკიდებულია ცხოველის საკვებისა და ცხოველის საკვებში ზეთის/ცხიმის ბუნებაზე. თავი IX ნედლი უჯრედანას განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა, რომ ცხოველის საკვებში განვსაზღვროთ უცხიმო ორგანული ნივთიერებები, რომლებიც არ იხსნება მჟავე და ტუტე გარემოში და ჩვეულებრივ აღწერილია, როგორც ნედლი უჯრედანა. 2. გამოყენების პრინციპი საჭიროების შემთხვევაში, ცხიმგამოცლილი ნიმუში მუშავდება თანმიმდევრულად გარკვეული კონცენტრაციის გოგირდმჟავასა და კაუსტიკური კალიუმის ხსნარებით დუღილის მდგომარეობაში. ნაშთის (ნარჩენის) განცალკავება წარმოებს ფილტრაციით გარეცხილი, გამომშრალი, შეწონილი და დანაცრებული სინთეზირებული მინის ფილტრის საშუალებით 475-დან 500°C ტემპერატურაზე. დანაცრების გამო წონის დანაკარგი შეესაბამება ნედლ უჯრედანას, რომელიც წარმოდგენილია გამოსაკვლევ ნიმუშში.
3. რეაგენტები 3.1. გოგირდმჟავა, c = 0,13 მოლი/ლ. 3.2. აქაფების საწინააღმდეგო საშუალება (აგენტი) (მაგ., n-ოქტანოლი). 3.3. ფილტრის საშუალებები (ცელიტი 545 ან ეკვივალენტი), თბება 500oC ტემპერატურაზე ოთხი საათის განმავლობაში (ამ თავის პუნქტი 8.6). 3.4. აცეტონი. 3.5. პეტროლეუმის ეთერის დუღილის დიაპაზონი 40-დან 60oC-მდე. 3.6. მარილმჟავა, c = 0,5 მოლი/ლ. 3.7. კალიუმის ჰიდროქსიდის ხსნარი, c = 0,23 მოლი/ლ.
4. აპარატურა 4.1. გამათბობელი მოწყობილობა გოგირდმჟავათი და კალიუმის ჰიდროქსიდის ხსნარით გადახარშვისთვის, რომელიც აღჭურვილია ფილტრაციის თიგელით (ამ თავის პუნქტი 4.2) და გამომავალი მილით, რომლის ბოლოში თავსახურია (ონკანი), სითხის გამოსაშვებად და ვაკუუმის შესაქმნელად, შესაძლოა აღჭურვილია კომპრესირებული ჰაერით. გამოყენებამდე ყოველდღე გააცხელეთ მოწყობილობა მდუღარე წყლით ხუთი წუთის განმავლობაში. 4.2. ფილტრაციის თიგელი (ფილტრი პერფორირებული ძირით), რომელიც შერწყმულია მინის ფილტრის ფირფიტის 40-90 მკმ ზომის მქონე ფორებთან (ნასვრეტებით). პირველ გამოყენებამდე, გააცხელეთ 500°C ტემპერატურაზე რამდენიმე წუთის განმავლობაში და შემდეგ გააგრილეთ (ამ თავის პუნქტი 8.6). 4.3. ცილინდრი სულ მცირე 270 მლ რეფლუქსის კონდენსატორით (უკუმაცივრით), შესაფერისი დუღილისთვის. 4.4. საშრობი ღუმელი თერმოსტატით. 4.5. მუფელური ღუმელი თერმოსტატით. 4.6. ექტრაჰირების მოწყობილობა, რომელიც შედგება ფილტრაციის თიგელისთვის (ამ თავის პუნქტი 4.2) საყრდენი ფირფიტისაგან, და ნარჩენების დამცლელი მილისაგან, რომელიც აღჭურვილია თავსახურით (ონკანით), სითხის გამოსაშვებად და ვაკუუმის შესაქმნელად. 4.7. დამაკავშირებელი რგოლები გამათბობელი ხელსაწყოს (ამ თავის პუნქტი 4.1), თიგელისა (ამ თავის პუნქტი 4.2) და ცილინდრის (ამ თავის პუნქტი 4.3) ასაწყობად და ცივი ექსტრაჰირების აპარატის (ამ თავის პუნქტი 4.6) და თიგელის დასაკავშირებლად.
5. გამოყენების პროცედურა აწონეთ 1 გ მომზადებული ნიმუში 1 მგ-მდე სიზუსტით და მოათავსეთ იგი თიგელში (ამ თავის პუნქტი 4.2), (იხ. ამ თავის პუნქტი 8.1, 8.2 და 8.3 დაკვირვებები) და დაამატეთ 1 გ გაფილტრული დამხმარე საშუალება (ადიუვანტი) (ამ თავის პუნქტი 3.3).
ააწყეთ გამათბობელი ხელსაწყო (ამ თავის პუნქტი 4.1) და ფილტრაციის თიგელი (ამ თავის პუნქტი 4.2), შემდეგ კი ცილინდრი მიამაგრეთ (ამ თავის პუნქტი 4.3) თიგელზე. ჩაასხით 150 მლ მდუღარე გოგირდმჟავა (ამ თავის პუნქტი 3.1) აწყობილ ცილინდრში და თიგელში და საჭიროების შემთხვევაში დაამატეთ რამდენიმე წვეთი აქაფების საწინააღმდეგო საშუალება (აგენტი) (ამ თავის პუნქტი 3.2).
სითხე მიიყვანეთ ადუღებამდე 5 ± 2 წუთის განმავლობაში და ძლიერად ადუღეთ ზუსტად 30 წუთი.
გახსენით ნარჩენების დამცლელი მილის ონკანი (ამ თავის პუნქტი 4.1) და, ვაკუუმის პირობებში, გაფილტრეთ გოგირდმჟავა ფილტრაციის თიგელის საშუალებით და შემდეგ ზედიზედ ჩამორეცხეთ ნაშთი (ნარჩენები) სამჯერ 30 მლ მდუღარე წყლით და დარწმუნდით, რომ ნაშთი (ნარჩენები) გაფილტრულია ყოველი გარეცხვის შემდეგ.
დახურეთ ნარჩენების დამცლელი მილის ონკანი და დაასხით 150 მლ მდუღარე კალიუმის ჰიდროქსიდის ხსნარი (ამ თავის პუნქტი 3.7) აწყობილ ცილინდრსა და თიგელში, და დაამატეთ რამდენიმე წვეთი აქაფების საწინააღმდეგო საშუალება (აგენტი) (ამ თავის პუნქტი 3.2). სითხე მიიყვანეთ დუღილის წერტილამდე 5 ± 2 წუთის განმავლობაში და ძლიერად ადუღეთ ზუსტად 30 წუთი. გაფილტრეთ და გაიმეორეთ გარეცხვის პროცედურა, რომელიც გამოიყენება გოგირდმჟავას საფეხურისთვის.
საბოლოო გარეცხვისა და გაშრობის შემდეგ, განაცალკევეთ თიგელი და მისი შემცველობა და ხელახლა შეუერთეთ ცივი ექსტრაჰირების ხელსაწყოს (ამ თავის პუნქტი 4.6). შექმენით ვაკუუმი და ჩამორეცხეთ ნარჩენები თიგელში ზედიზედ 25 მლ აცეტონის პორციებით (ამ თავის პუნქტი 3.4), თან დარწმუნდით, რომ ნაშთი (ნარჩენები) გაფილტრულია ყოველი ჩამორეცხვის შემდეგ.
მუდმივი წონის მისაღებად თიგელი უნდა გააშროთ ღუმელში 130°C ტემპერატურაზე. ყოველი გაშრობის შემდეგ გააგრილეთ დესიკატორში (საშრობში) და სწრაფად აწონეთ. თიგელი მოათავსეთ მუფელურ ღუმელში და მოახდინეთ მისი დანაცრება მუდმივი წონის მისაღწევად (წონის დანაკარგი ორ თანმიმდევრულ წონას შორის უნდა იყოს 2 მგ-ზე ნაკლები ან ტოლი) 475oC-დან 500oC ტემპერატურამდე, სულ მცირე 30 წუთის განმავლობაში.
ყოველი გათბობის შემდეგ ჯერ გააცივეთ ღუმელში, შემდეგ კი დესიკატორში (საშრობში) აწონვამდე.
განახორციელეთ ბრმა (საკონტროლო) ტესტი ნიმუშის გარეშე. დანაცრების შედეგად წონის დანაკარგი არ უნდა აღემატებოდეს 4 მგ-ს.
6. შედეგების დაანგარიშება ნედლი უჯრედანას შემცველობა, როგორც ნიმუშის პროცენტული მაჩვენებელი, მოცემულია შემდეგი ფორმულით:
სადაც: m = ნიმუშის წონა გ-ში; m0 = დანაცრების შემდეგ წონის დანაკარგი განსაზღვრის დროს, გ-ში; m1 = დანაცრების შემდეგ წონის დანაკარგი ბრმა (საკონტროლო) ტესტის დროს, გ-ში.
7. განმეორებადობა ერთ ნიმუშზე განხორციელებულ ორ პარალელურ განსაზღვრას შორის სხვაობა არ უნდა აღემატებოდეს: – 0,6%-ს აბსოლუტურ მნიშვნელობაში, ნედლი ბოჭკოვანას შემცველობისთვის 10% -ზე ქვემოთ; – 6%-ს შედარებით მაღალ შედეგთან მიმართებაში, ნედლი ბოჭკოვანას 10%-ზე მეტი ან ტოლი შემცველობისთვის.
8. დაკვირვება 8.1. ცხოველის საკვებიდან, რომელიც შეიცავს 10% -ზე მეტ ნედლ ცხიმს, უნდა მოხდეს ცხიმის მოცილება პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.5).
ფილტრაციის თიგელი (ამ თავის პუნქტი 4.2) და მისი შემცველობა მიუერთეთ ცივი ექსტრაჰირების ხელსაწყოს (ამ თავის პუნქტი 4.6) და შექმენით ვაკუუმი, და თიგელში არსებული ნაშთი (ნარჩენები) ჩამორეცხეთ ზედიზედ სამჯერ 30 მლ პეტროლეუმის ეთერით, თან დარწმუნდით, რომ ნაშთი (ნარჩენები) მშრალია. თიგელი და მისი შემცველობა შეუერთეთ გამათბობელ მოწყობილობას (ამ თავის პუნქტი 4.1) და გააგრძელეთ ამ თავის მე-5 პუნქტის შესაბამისად. 8.2. ცხოველის ცხიმშემცველი იმ საკვებისგან, რომელთა ექსტრაჰირება შეუძლებელია უშუალოდ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.5), უნდა მოხდესა მათგან ცხიმის გამოცლა ამ თავის 8.1 პუნქტის შესაბამისად და კიდევ ერთხელ უნდა მოხდეს ცხიმის გამოცლა მჟავასთან ერთად ადუღების შემდეგაც. მჟავასთან ერთად დუღილისა და შემდგომი ჩამორეცხვის შემდეგ თიგელი და მისი შემცველობა დააკავშირეთ ცივი ექსტრაჰირების ხელსაწყოსთან (ამ თავის პუნქტი 4.6) და სამჯერ გარეცხეთ 30 მლ აცეტონით, რის შემდეგ კი გარეცხეთ სამჯერ 30 მლ პეტროლეუმის ეთერის პორციებით. გაფილტეთ ვაკუუმში, სანამ არ გაშრება და გააგრძელეთ გამოკვლევა, როგორც ეს აღწერილია ამ თავის მე-5 პუნქტში, და გამოკვლევა დაიწყეთ კალიუმის ჰიდროქსიდის დამუშავებით. 8.3. თუ ცხოველი საკვები შეიცავს 5%-ზე მეტ ნახშირწყლებს, რომელიც გამოხატულია კალციუმის კარბონატად, შეაერთეთ ფილტრაციის თიგელი (ამ თავის პუნქტი 4.2) აწონილ ნიმუშთან ერთად გამათბობელ ხელსაწყოსთან (ამ თავის პუნქტი 4.1). ნიმუში სამჯერ გარეცხეთ 30 მლ მარილმჟავათი (ამ თავის პუნქტი 3.6). ყოველი დამატების შემდეგ ნიმუში გააჩერეთ ფილტრაციის დაწყებამდე დაახლოებით ერთი წუთით. ერთხელ გარეცხეთ 30 მლ წყლით და შემდეგ გააგრძელეთ, როგორც ეს აღწერილია ამ თავის მე-5 პუნქტში. 8.4. თუკი აპარატი/დანადგარი გამოიყენება საყრდენის სახით (რამდენიმე თიგელი მიერთებულია ერთსა და იმავე გამათბობელ ხელსაწყოზე), ორი ინდივიდუალური განსაზღვრის ჩატარება არ შეიძლება ერთსა და იმავე გამოსაკვლევ ნიმუშზე ერთსა და იმავე სერიაში. 8.5. თუ დუღილის შემდეგ ძნელია მჟავას და ძირითადი ხსნარების გაფილტვრა, გამოიყენეთ კომპრესირებული ჰაერი, რომელიც შეჰყავთ გამათბობელი ხელსაწყოს დამცლელი მილის საშუალებით და შემდეგ გააგრძელეთ ფილტრაცია. 8.6. დანაცრების ტემპერატურა არ უნდა აღემატებოდეს 500°C-ს, რათა მინის ფილტრაციის თიგელის გამოყენება გახანგრძლივდეს. გათბობისა და გაგრილების ციკლების დროს საჭიროა სიფრთხილის გამოჩენა, რათა თავიდან იქნეს აცილებული ზედმეტი თერმული შოკი. თავი X შაქრის განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი შესაძლებლობას გვაძლევს, ინვერსიის შემდეგ განვსაზღვროთ შაქრის აღდგენილი და საერთო რაოდენობა, რომელიც გამოხატულია, როგორც გლუკოზა, და საჭიროების შემთხვევაში, როგორც საქაროზა, 0,95 ფაქტორით (კოეფიციენტის) გარდაქმნისას. იგი გამოიყენება ცხოველის კომბინირებულ საკვებთან მიმართებაში. ცხოველის სხვა საკვებისთვის გათვალისწინებულია სპეციალური მეთოდები. საჭიროების შემთხვევაში ლაქტოზა იზომება ცალკე და მხედველობაში მიიღება შედეგების გაანგარიშებისას.
2. გამოყენების პრინციპი შაქარის ექსტრაჰირება წარმოებს განზავებულ ეთანოლში; ხსნარი სუფთავდება Carrez-ის I და II ხსნარით. ეთანოლის ექსტრაჰირების შემდეგ რაოდენობა, ინვერსიის დაწყებამდე და შემდეგ განისაზღვრება Luff-Schoorl-ის მეთოდით.
3. რეაგენტები 3.1. ეთანოლის ხსნარი 40% (მ/მ) სიმკვრივე: 0,948 გ/მლ 20°C ტემპერატურაზე, ნეიტრალიზდება ფენოლფთალინით. 3.2. Carrez-ის I ხსნარი: წყალში გახსენით 21,9 გ თუთიის აცეტატი Zn(CH3COO)22H2O და 3 გ დაკრისტალებული ძმარმჟავა. შეავსეთ 100 მლ-მდე წყლით. 3.3. Carrez-ის ხსნარი II: წყალში გახსენით 10,6 გ კალიუმის ფეროციანიდი K4Fe(CN)6 3H2O. შეავსეთ 100 მლ წყლით. 3.4. მეთილ-ნარინჯი, ხსნარი 0,1% (წონ/მოც). 3.5. მარილმჟავა 4 მოლი/ლიტრი. 3.6. მარილმჟავა 0,1 მოლი/ლიტრი. 3.7. ნატრიუმის ჰიდროქსიდის ხსნარი 0,1 მოლი/ლიტრი. 3.8. Luff-Schoorl-ის რეაგენტი: ფრთხილად აურიეთ, დაასხით ლიმონმჟავას ხსნარი (ამ თავის პუნქტი 3.8.2) ნატრიუმის კარბონატის ხსნარში (ამ თავის პუნქტი 3.8.3). დაამატეთ სპილენძის სულფატის ხსნარი (ამ თავის პუნქტი 3.8.1) და შეავსეთ 1 ლიტრამდე წყლით. დატოვეთ დასალექად მთელი ღამე და გაფილტრეთ. შეამოწმეთ ამგვარად მიღებული რეაგენტის კონცენტრაცია (Cu 0,05 მოლი/ლიტრი; Na2CO3 1 მოლი/ლიტრი), იხილეთ ამ თავის 5.4 პუნქტის ბოლო ქვეპუნქტი. ხსნარის pH უნდა იყოს დაახლოებით 9,4. 3.8.1. სპილენძის სულფატის ხსნარი: 25 გ სპილენძის სულფატი CuSO45H2O, რკინისგან თავისუფალი, გახსენით 100 მლ წყალში. 3.8.2. ლიმონმჟავას ხსნარი: 50 გ ლიმონმჟავა C6H8O7·H2O, გახსენით 50 მლ წყალში. 3.8.3. ნატრიუმის კარბონატის ხსნარი: გახსენით 143,8 გ უწყლო ნატრიუმის კარბონატი 300 მლ თბილ წყალში. დატოვეთ გასაგრილებლად. 3.9. ნატრიუმის თიოსულფატის ხსნარი 0,1 მოლი/ლიტრი. 3.10. სახამებლის ხსნარი: 1 ლიტრ მდუღარე წყალში დაამატეთ 5 გ ხსნადი სახამებლის ნარევი 30 მლ წყლით. ადუღეთ სამი წუთის განმავლობაში, გააჩერეთ გასაგრილებლად და საჭიროების შემთხვევაში დაამატეთ 10 მგ ვერცხლისწყლის იოდიდი, როგორც კონსერვანტი. 3.11. გოგირდმჟავა 3 მოლი/ლიტრი. 3.12. კალიუმის იოდიდი, ხსნარი 30% (წ/მ). 3.13. გრანულირებული პემზის ქვა მოხარშულია მარილმჟავაში, გარეცხილია წყალში და გამშრალია. 3.14. 3-მეთილბუტან-l-ოლი.
4. აპარატი მიქსერი (ტუმბლერი): დაახლოებით 35-დან 40 ბრ./წთ.
5. მომზადების პროცედურა
5.1 ნიმუშის ექსტრაჰირება აწონეთ 2,5 გ ნიმუში მგ-მდე სიზუსტით და მოათავსეთ 250 მლ მოცულობის კოლბაში. დაამატეთ 200 მლ ეთანოლი (ამ თავის პუნქტი 3.1) და ურიეთ შემრევში (ტუმბლერში) ერთი საათის განმავლობაში. დაამატეთ 5 მლ Carrez-ის ხსნარი I (ამ თავის პუნქტი 3.2) და ურიეთ დაახლოებით 30 წამი. დაამატეთ 5 მლ Carrez-ის ხსნარი II (ამ თავის პუნქტი 3.3) და კვლავ ურიეთ ერთი წუთის განმავლობაში. შეავსეთ მოცულობა ეთანოლით (ამ თავის პუნქტი 3.1), მოახდინეთ ჰომოგენიზაცია და გაფილტრეთ. ამოიღეთ 200 მლ ფილტრატი და ააორთქლეთ დაახლოებით ნახევარ მოცულობამდე, რათა აღმოიფხვრას ეთანოლის უმეტესი ნაწილი. აორთქლების ნაშთი (ნარჩენები) რაოდენობრივად გადაიტანეთ 200 მლ მოცულობის კოლბაში თბილი წყლის გამოყენებით, გააგრილეთ, შეავსეთ წყლით საჭირო მოცულობამდე, მოახდინეთ ჰომოგენიზაცია და საჭიროების შემთხვევაში გაფილტრეთ. ეს ხსნარი გამოყენებული იქნება შაქრის აღდგენილი რაოდენობის და, ინვერსიის შემდეგ, შაქრის საერთო რაოდენობის განსაზღვრისთვის.
5.2. აღდგენილი შაქრის განსაზღვრა პიპეტის გამოყენებით ამოიღეთ არაუმეტეს 25 მლ ხსნარი, რომელიც შეიცავს 60 მგ-ზე ნაკლებ აღდგენილ შაქარს, გამოხატულს გლუკოზით. საჭიროების შემთხვევაში შეავსეთ 25 მლ-მდე გამოხდილი წყლით და განსაზღვრეთ შაქრის აღდგენილი რაოდენობა Luff-Schoorl მეთოდით. შედეგი გამოიხატება როგორც გლუკოზის პროცენტული მაჩვენებელი ნიმუშში.
5.3. ინვერსიის შემდეგ შაქრის საერთო შემცველობის განსაზღვრა პიპეტის გამოყენებით აიღეთ 50 მლ ხსნარი და გადაიტანეთ 100 მლ მოცულობის კოლბაში. დაამატეთ მეთილ-ნარინჯის რამდენიმე წვეთი (ამ თავის პუნქტი 3.4). შემდეგ ფრთხილად და შეუჩერებლად ურიეთ, დაუმატეთ მარილმჟავა (ამ თავის პუნქტი 3.5), ვიდრე სითხე არ მიიღებს კარგად გამოხატულ წითელ ფერს. დაუმატეთ 15 მლ მარილმჟავა (ამ თავის პუნქტი 3.6), მოათავსეთ კოლბა სწრაფად მოდუღარე წყლის აბაზანაში და დააყოვნეთ 30 წუთის განმავლობაში. სწრაფად გააგრილეთ დაახლოებით 20°C ტემპერატურამდე და დაუმატეთ 15 მლ ნატრიუმის ჰიდროქსიდის ხსნარი (ამ თავის პუნქტი 3.7). შეავსეთ 100 მლ-მდე წყლით და მოახდინეთ ჰომოგენიზაცია. ამოიღეთ არაუმეტეს 25 მლ, რომელიც შეიცავს 60 მგ-ზე ნაკლებ აღდგენილ შაქარს, გამოხატულს როგორც გლუკოზას. საჭიროების შემთხვევაში შეავსეთ 25 მლ-მდე გამოხდილი წყლით და განსაზღვრეთ შაქრის აღდგენილი რაოდენობა Luff-Schoorl მეთოდით. შედეგი გამოიხატება გლუკოზის პროცენტულ მაჩვენებლებში, საჭიროების შემთხვევაში, საქაროზის პროცენტებში, 0,95 ფაქტორზე (კოეფიციენტის) გამრავლების გზით.
5.4. ტიტრაცია Luff-Schoorl-ის მეთოდით პიპეტის გამოყენებით აიღეთ 25 მლ Luff-Schoorl რეაგენტი (ამ თავის პუნქტი 3.8) და გადაიტანეთ 300 მლ ერლენმაიერის კოლბაში; დაამატეთ ზუსტად 25 მლ გაწმენდილი შაქრის ხსნარი. დაამატეთ 2 გრანულირებული პემზის ქვა (ამ თავის პუნქტი 3.13), გააცხელეთ, ხელით ურიეთ საშუალო სიმაღლის ღია ალზე და სითხე მიიყვანეთ დუღილამდე დაახლოებით ორ წუთში. ერლენმაიერი დაუყოვნებლივ მოათავსეთ აზბესტით დაფარულ მავთულის ბადეზე, რომლის დიამეტრი დაახლოებით 6 სმ სიგრძისაა და რომლის ქვემოთაც ალია ანთებული. ალი უნდა დარეგულირდეს ისე, რომ მხოლოდ ერლენმაიერის ძირი გახურდეს. ერლენმაიერის კოლბაში დაამონტაჟეთ უკუმაცივარი (რეფლუქსის კონდენსატორი). ადუღეთ ზუსტად 10 წუთი. დაუყოვნებლივ გააგრილეთ ცივ წყალში და დაახლოებით ხუთი წუთის შემდეგ გატიტრეთ შემდეგნაირად:
დაამატეთ 10 მლ კალიუმის იოდიდის ხსნარი (ამ თავის პუნქტი 3.12) და ამის შემდეგ დაუყოვნებლივ (ფრთხილად, გადაჭარბებული ქაფის მიღების რისკის გამო) დაამატეთ 25 მლ გოგირდმჟავა (ამ თავის პუნქტი 3.11). გატიტრეთ ნატრიუმის თიოსულფატის ხსნარით (ამ თავის პუნქტი 3.9), ვიდრე არ გამოჩნდება მკრთალი ყვითელი ფერი, დაამატეთ სახამებლის ინდიკატორი (ამ თავის პუნქტი 3.10) და დაასრულეთ ტიტრაცია.
იმავე ტიტრაცია ჩაატარეთ 25 მლ Luff-Schoorl-ის რეაგენტის (ამ თავის პუნქტი 3.8) და 25 მლ წყლის ზუსტად გაზომილ ნარევზე, 10 მლ კალიუმის იოდიდის ხსნარისა (ამ თავის პუნქტი 3.12) და 25 მლ გოგირდმჟავას (ამ თავის პუნქტი 3.11) დამატების შემდეგ, დუღილის გარეშე.
6. შედეგების დაანგარიშება ცხრილის საშუალებით დაადგინეთ გლუკოზის რაოდენობა მგ-ში, რომელიც შეესაბამება სხვაობას ორი ტიტრაციის მნიშვნელობას შორის, გამოხატულს მგ-ში ნატრიუმის თიოსულფატის 0,1 მოლი/ლიტრზე. შედეგი გამოხატეთ როგორც ნიმუშის პროცენტული მაჩვენებელი.
7. სპეციალური პროცედურები 7.1. ცხოველის საკვების შემთხვევაში, რომლებიც მდიდარია ბადაგითა და ცხოველის სხვა საკვებით, რომლებიც არ არის განსაკუთრებით ჰომოგენიზებული, აწონეთ 20 გ და მოათავსეთ 500 მლ წყლით 1 ლიტრის მოცულობის კოლბაში. ურიეთ ერთი საათის განმავლობაში შემრევში (ტუმბულში). გაწმინდეთ Carrez I (ამ თავის პუნქტი 3.2) და II (ამ თავის პუნქტი 3.3) რეაგენტების გამოყენებით ისე, როგორც ეს აღწერილია ამ თავის 5.1 პუნქტში, ამჯერად კი გამოიყენეთ თითოეულ რეაქტივზე ოთხჯერ მეტი რაოდენობა. შეავსეთ მოცულობა 80% ეთანოლით (მოც/მოც).
მოახდინეთ ჰომოგენიზაცია და გაფილტრეთ. აღმოფხვერით ეთანოლი, როგორც ეს აღწერილია ამ თავის 5.1 პუნქტში. თუ დექსტრინიზებული სახამებელი არ არის, შეავსეთ მოცულობა გამოხდილი წყლით. 7.2. ბადაგისა და ცხოველის საკვების მასალების შემთხვევაში, რომლებიც მდიდარია შაქრით და თითქმის თავისუფალია სახამებლისგან (კერატი, გამომშრალი ჭარხლის კასეტები და ა.შ.), აწონეთ 5 გრ, მოათავსეთ 250 მლ მოცულობის კოლბაში, დაამატეთ 200 მლ გამოხდილი წყალი და ურიეთ შემრევი (ტუმბო) ერთი საათის განმავლობაში ან მეტი დროით, საჭიროების შემთხვევაში. გაწმინდეთ Carrez I (ამ თავის პუნქტი 3.2) და II (ამ თავის პუნქტი 3.3) რეაგენტების გამოყენებით, როგორც ეს აღწერილია ამ თავის 5.1 პუნქტში. შეავსეთ მოცულობამდე ცივი წყლით, მოახდინეთ ჰომოგენიზაცია და გაფილტვრა. შაქრის საერთო ოდენობის განსაზღვრისთვის, განაგრძეთ ისე, როგორც ეს აღწერილია ამ თავის 5.3 პუნქტში.
8. დაკვირვება 8.1. აქაფების თავიდან ასაცილებლად, სასურველია დაემატოს (მოცულობის მიუხედავად) დაახლოებით 1 მლ 3-მეთილბუტან-l-ოლი (ამ თავის პუნქტი 3.14), სანამ აადუღებთ Luff-Schoorl-ის რეაგენტთან. 8.2. ინვერსიის შემდეგ შაქრის საერთო შემცველობებს შორის განსხვავება, გამოხატული როგორც გლუკოზა და აღდგენილი შაქრის შემცველობა, გამოხატული როგორც გლუკოზა, მრავლდება 0,95-ზე, რაც იძლევა საქაროზის პროცენტულ შემცველობას. 8.3. აღდგენილი შაქრის შემცველობის განსასაზღვრად, ლაქტოზას გარდა, შეიძლება გამოყენებულ იქნეს ორი მეთოდი: 8.3.1. მიახლოებითი გაანგარიშებისთვის, გაამრავლეთ 0,675-ზე ლაქტოზას შემცველობა, რომელიც დადგენილია გამოკვლევის სხვადასხვა მეთოდით და გამოაკელით აღდგენილი შაქრის შემცველობიდან მიღებული შედეგი. 8.3.2. აღდგენილი შაქრის ზუსტი გაანგარიშებისთვის, ლაქტოზას გარდა, იმავე ნიმუში უნდა იქნეს გამოყენებული ორი საბოლოო განსაზღვრისთვის. ერთ-ერთი გამოკვლევა ხორციელდება ამ თავის 5.1 პუნქტით გათვალისწინებული ხსნარის ნაწილზე, მეორე კი, ლაქტოზას განსაზღვრის დროს მიღებული ხსნარის ნაწილზე, ამ მიზნით დადგენილი მეთოდით (სხვა სახის შაქრის ფერმენტაციისა და გაწმენდის შემდეგ).
ორივე შემთხვევაში წარმოდგენილი შაქრის რაოდენობა განისაზღვრება Luff-Schoorl-ის მეთოდით და გამოითვლება გლუკოზას მგ-ში. ერთ-ერთი მნიშვნელობა გამოაკლდება სხვას და განსხვავება გამოიხატება, როგორც ნიმუშის პროცენტული მაჩვენებელი.
მაგალითი: მიღებული ორი მნიშვნელობა შეესაბამება, თითოეულ განსაზღვრისთვის, ნიმუშის 250 მგ-ს. პირველ შემთხვევაში მოიხმარს 17 მლ ნატრიუმის თიოსულფატის ხსნარს 0,1 მოლი/ლიტრზე, რომელიც შეესაბამება 44,2 მგ გლუკოზას; მეორეში, 11 მლ, რაც შეესაბამება 27,6 მგ გლუკოზას.
განსხვავება არის 16,6 მგ გლუკოზა.
აღდგენილი შაქრის შემცველობა (ლაქტოზას გარდა), რადგან გამოანგარიშებულია როგორც გლუკოზა, არის შესაბამისად:
25 მლ Luff-Schoorl-ის რეაგენტისთვის მნიშვნელობათა ცხრილი Na2S2O3 მლ 0,1 მოლი/ლიტრი, გათბობა ორი წუთის განმავლობაში, დუღილი 10 წუთი
თავი XI ლაქტოზას განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას გვაძლევს განვსაზღვროთ ლაქტოზას რაოდენობა ცხოველის საკვებში, რომელიც შეიცავს 0,5%-ზე მეტ ლაქტოზას. 2. გამოყენების პრინციპი შაქარი იხსნება წყალში. ხსნარი ექვემდებარება ფერმენტაციას, Saccharomyces cerevisiae საფუარით, რომელიც ტოვებს ლაქტოზას ხელუხლებელს. გაწმენდისა და ფილტრაციის შემდეგ ლაქტოზას შემცველობა ფილტრატში განისაზღვრება Luff-Schoorl-ის მეთოდით. 3. რეაგენტები 3.1. Saccharomyces cerevisiae სუსპენზია: გახსენით 25 გ ახალი საფუარი 100 მლ წყალში. სუსპენზია შეიძლება შევინახოთ მაცივარში მაქსიმუმ ერთი კვირის განმავლობაში. 3.2. Carrez-ის ხსნარი I: წყალში გახსენით 21,9 გ თუთიის აცეტატი, Zn (CH3COO)2 2H2O და 3 გ დაკრისტალებული ძმარმჟავა. შეავსეთ 100 მლ-მდე წყლით. 3.3. Carrez-ის ხსნარი II: წყალში გახსენით 10,6 გ კალიუმის ფეროციანიდი K4Fe (CN)6 3H2O. შეავსეთ 100 მლ წყლით. 3.4. Luff-Schoorl-ის რეაგენტი: ფრთხილად აურიეთ, ჩაასხით ლიმონმჟავას ხსნარი (ამ თავის პუნქტი 3.8.2) ნატრიუმის კარბონატის ხსნარში (ამ თავის პუნქტი 3.4.3). დაამატეთ სპილენძის სულფატის ხსნარი (ამ თავის პუნქტი 3.8.1) და შეავსეთ 1 ლიტრამდე წყლით. გააჩერეთ დასალექად მთელი ღამე და გაფილტრეთ. შეამოწმეთ ამგვარად მიღებული რეაგენტის კონცენტრაცია (Cu 0,05 მოლი/ლიტრი; Na2CO3 1 მოლი/ლიტრი), იხილეთ ამ თავის 5.4-ის ბოლო ქვეპუნქტი. ხსნარის pH უნდა იყოს დაახლოებით 9,4. 3.4.1. სპილენძის სულფატის ხსნარი: 25 გ სპილენძის სულფატი CuSO45H2O, რკინის გარეშე, გახსენით 100 მლ წყალში. 3.4.2. ლიმონმჟავას ხსნარი: 50 გ ლიმონმჟავა C6H8O7 · H2O, გახსენით 50 მლ წყალში. 3.4.3. ნატრიუმის კარბონატის ხსნარი: გახსენით 143,8 გ უწყლო ნატრიუმის კარბონატი 300 მლ თბილ წყალში. დატოვეთ გასაგრილებლად. 3.5. გრანულირებული პემზის ქვა მოხარშულია მარილმჟავაში, წყალშია გარეცხილი და გამშრალია. 3.6. კალიუმის იოდიდი, ხსნარი 30% (წ/მ). 3.7. გოგირდმჟავა 3 მოლი/ლიტრი. 3.8. ნატრიუმის თიოსულფატის ხსნარი 0,1 მოლი/ლიტრზე. 3.9. სახამებლის ხსნარი: 1 ლიტრ მდუღარე წყალში დაამატეთ 5 გ ხსნადი სახამებლის ნარევი 30 მლ წყალით. ადუღეთ სამი წუთის განმავლობაში, გააჩერეთ გასაგრილებლად და საჭიროების შემთხვევაში დაამატეთ 10 მგ ვერცხლისწყლის იოდიდი, როგორც კონსერვანტი. 4. აპარატურა 38-40oC ტემპერატურაზე დაყენებულ თერმოსტატზე წყლის აბაზანა 5. მომზადების პროცედურა აწონეთ 1 გ ნიმუში მგ-მდე სიზუსტით და მოათავსეთ ნიმუშის ეს პორცია 100 მლ მოცულობის კოლბაში. დაამატეთ 25-დან 30 მლ-მდე წყალი. კოლბა მოათავსეთ მდუღარე წყლის აბაზანაში 30 წუთის განმავლობაში და შემდეგ გააგრილეთ დაახლოებით 35°C-მდე. დაამატეთ 5 მლ საფუარის სუსპენზია (ამ თავის პუნქტი 3.1) და მოახდინეთ ჰომოგენიზაცია. კოლბა გააჩერეთ ორი საათის განმავლობაში წყლის აბაზანაში, 38-40°C ტემპერატურაზე. გააგრილეთ დაახლოებით 20°C-მდე.
დაამატეთ 2,5 მლ Carrez-ის ხსნარი I (ამ თავის პუნქტი 3.2) და ურიეთ 30 წამი, შემდეგ დაამატეთ 2,5 მლ Carrez-ის ხსნარი II (ამ თავის პუნქტი 3.3) და კვლავ ურიეთ 30 წამის განმავლობაში. შეავსეთ 100 მლ-მდე წყლით, აურიეთ და გაფილტრეთ. პიპეტის გამოყენებით ამოიღეთ 40-დან 80-მდე მგ ლაქტოზას ოდენობა და გადაიტანეთ 300-მლ-იან ერლენმაიერის კოლბაში. საჭიროების შემთხვევაში, შეავსეთ 25 მლ წყლით.
ჩაატარეთ საკონტროლო ტესტი ანალოგიურად 5 მლ საფუარის სუსპენზიით (ამ თავის პუნქტი 3.1). განსაზღვრეთ ლაქტოზას შემცველობა Luff-Schoorl-ის შესაბამისად, შემდეგნაირად: დაამატეთ ზუსტად 25 მლ Luff-Schoorl-ის რეაგენტი (ამ თავის პუნქტი 3.4) და ორი გრანულირებული პემზის ქვა (ამ თავის პუნქტი 3.5). ხელით ურიეთ საშუალო სიმაღლის ღია ალზე და სითხე მიიყვანეთ დუღილამდე დაახლოებით ორ წუთის განმავლობაში. ერლენმაიერი დაუყოვნებლივ მოათავსეთ აზბესტით დაფარულ მავთულის ბადეზე, რომლის ნასვრეტის დიამეტრი დაახლოებით 6 სმ სიგრძისაა და რომლის ქვემოთაც ანთებულია ალი. ალი უნდა დარეგულირდეს ისე, რომ მხოლოდ ერლენმაიერის ძირი გახურდეს. ერლენმაიერის კოლბაში დაამონტაჟეთ უკუმაცივარი (რეფლუქსის კონდენსატორი). ადუღეთ ზუსტად 10 წუთი. გააგრილდეთ დაუყოვნებლივ ცივ წყალში და დაახლოებით ხუთი წუთის შემდეგ გატიტრეთ შემდეგნაირად:
დაამატეთ 10 მლ კალიუმის იოდიდის ხსნარი (ამ თავის პუნქტი 3.6) და ამის შემდეგ დაუყოვნებლივ (ფრთხილად, გადაჭარბებული ქაფის მიღების რისკის გამო), დაამატეთ 25 მლ გოგირდმჟავა (ამ თავის პუნქტი 3.7). გატიტრეთ ნატრიუმის თიოსულფატის ხსნარით (ამ თავის პუნქტი 3.8) სანამ არ გამოჩნდება მკრთალი ყვითელი ფერი, დაამატეთ სახამებლის ინდიკატორი (ამ თავის პუნქტი 3.9) და დაასრულეთ ტიტრაცია.
იმავე ტიტრაცია ჩაატარეთ 25 მლ Luff-Schoorl-ის რეაგენტის (ამ თავის პუნქტი 3.8) და 25 მლ წყლის ზუსტად გაზომილ ნარევზე, 10 მლ კალიუმის იოდიდის ხსნარისა (ამ თავის პუნქტი 3.6) და 25 მლ გოგირდმჟავას (ამ თავის პუნქტი 3.11) დამატების შემდეგ, დუღილის გარეშე.
6. შედეგების დაანგარიშება თანდართული ცხრილის საშუალებით დაადგინეთ ლაქტოზას რაოდენობა მგ-ში, რომელიც შეესაბამება სხვაობას ორი ტიტრაციის მნიშვნელობას შორის, გამოხატულს მგ-ში ნატრიუმის თიოსულფატის 0,1 მოლი/ლიტრზე. უწყლო ლაქტოზას შედეგი გამოხატეთ როგორც ნიმუშის პროცენტული მაჩვენებელი. 7. დაკვირვება პროდუქტებისთვის, რომლებიც შეიცავენ ფრეგმენტირებული შაქარის 40%-ზე მეტს, გამოიყენეთ 5 მლ-ზე მეტი საფუარის სუსპენზია (ამ თავის პუნქტი 3.1).
25 მლ Luff-Schoorl-ის რეაგენტისთვის მნიშვნელობათა ცხრილი Na2S2O3 მლ 0,1 მოლი/ლიტრი, გათბობა ორ წუთის განმავლობაში, დუღილი 10 წუთი
თავი XII სახამებლის განსაზღვრა – პოლარიმეტრული მეთოდი
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ სახამებლისა და მაღალ მოლეკულური წონის სახამებლის დეგრადაციის პროდუქტების შემცველობა ცხოველის საკვებში, რათა დადგენილ (შემოწმებულ) იქნეს დეკლარირებულ ენერგეტიკულ ღირებულებასთან (ამ წესის დანართი №7) და „ცხოველის საკვების გამოყენებისა და ბაზარზე განთავსების წესის დამტკიცების შესახებ“ საქართველოს მთავრობის 2021 წლის 8 თებერვლის №58 დადგენილებით განსაზღვრულ მოთხოვნებთან შესაბამისობა. 2. გამოყენების პრინციპი მეთოდი მოიცავს ორ განსაზღვრას. პირველში, ნიმუში დამუშავებულია განზავებული მარილმჟავათი. გაწმენდისა და ფილტრაციის შემდეგ ხსნარის ოპტიკური ბრუნვა იზომება პოლარიმეტრიით.
მეორეში, ნიმუში ექსტრაჰირებულია 40% ეთანოლით. ფილტრატის მარილმჟავათი აციდიფიკაციის, გაწმენდისა და გაფილტვრის შემდეგ იზომება ოპტიკური ბრუნვა (როტაცია), როგორც პირველი განსაზღვრისას.
განსხვავება ორ გაზომვას შორის, რომელიც მრავლდება ცნობილ ფაქტორზე, იძლევა ნიმუშის სახამებლის შემცველობას.
3. რეაგენტები 3.1. მარილმჟავა, ხსნარი 25% (წ/წ) სიმკვრივე: 1,126 გ/მლ. 3.2. მარილმჟავა. ხსნარი 1,13% (წ/მ) კონცენტრაცია უნდა შემოწმდეს ტიტრაციით ნატრიუმის ჰიდროქსიდის ხსნარის გამოყენებით 0,1 მოლი/ლიტრზე, 94% (მ/მ) ეთანოლში 0,1% (წ/მ) მეთილის წითელის თანდასწრებით. 10 მლ ნეიტრალიზაციისთვის საჭიროა 30,94 მლ NaOH 0,1 მოლი/ლიტრი. 3.3. Carrez-ის ხსნარი I: წყალში გახსენით 21,9 გ თუთიის აცეტატი Zn (CH3COO)22H2O და 3 გ დაკრისტალებული ძმარმჟავა. შეავსეთ 100 მლ-მდე წყლით. 3.4. Carrez-ის ხსნარი II: წყალში გახსენით 10,6 გ კალიუმის ფეროციანიდი K4Fe(CN)63H2O. შეავსეთ 100 მლ-მდე წყლით. 3.5. ეთანოლი, ხსნარი 40% (მ/მ), სიმკვრივე: 0,948 გ/მლ 20°C ტემპერატურაზე.
4. აპარატურა 4.1. 250 მლ ერლენმეიერის კოლბა სტანდარტული გლუვი (ხორკლიანი) მინით და უკუმაცივრით (რეფლუქსის კონდენსატორით). 4.2. პოლარიმეტრი ან სახარიმეტრი.
5. მომზადების პროცედურა
5.1. ნიმუშის მომზადება დააქუცმაცეთ ნიმუში იქამდე ვიდრე არ მიიღწევა ის ზომა, რომლითაც შესაძლებელი იქნება მისი მთლიანი გატარება 0.5 მმ-იანი ხვრელების მქონე მგრგვალ საცერში.
5.2. მთლიანი ოპტიკური ბრუნვის (როტაციის) განსაზღვრა (P ან S) (იხ. ამ თავის პუნქტი 7.1 -დაკვირვება) აწონეთ დაქუცმაცებული ნიმუში 2,5 გ მგ-მდე სიზუსტით და მოათავსეთ 100 მლ გრადირებულ (დანაყოფებიან) კოლბაში. დაამატეთ 25 მლ მარილმჟავა (ამ თავის პუნქტი 3.2), შეანჯღრიეთ, რომ მიიღოთ გამოსაკვლევი ნიმუშის თანაბარი განაწილება და დაამატეთ კიდევ 25 მლ მარილმჟავა (ამ თავის პუნქტი 3.2). კოლბა ჩაუშვით მდუღარე წყლის აბაზანაში, თან შეანჯღრიეთ ენერგიულად და შეუჩერებლივ პირველი სამი წუთის განმავლობაში აგლომერატების წარმოქმნის თავიდან ასაცილებლად. წყლის აბაზანაში წყლის რაოდენობა უნდა იყოს საკმარისი იმისათვის, რომ აბაზანამ შეინარჩუნოს დუღილის წერტილი, როდესაც მასში ვდებთ კოლბას. შეჯანჯღარებისას კოლბა არ უნდა ამოვიღოთ აბაზანიდან. ზუსტად 15 წუთის შემდეგ, კოლბა ამოიღეთ აბაზანიდან, დაამატეთ 30 მლ ცივი წყალი და გააცივეთ დაუყოვნებლივ 20oC ტემპერატურამდე.
დაამატეთ 5 მლ Carrez-ის ხსნარი I (ამ თავის პუნქტი 3.3) და შეანჯღრიეთ დაახლოებით 30 წამით. შემდეგ დაამატეთ 5 მლ Carrez -ისხსნარი II (ამ თავის პუნქტი 3.4) და კვლავ შეანჯღრიეთ დაახლოებით 30 წამით. შეავსეთ მოცულობა წყლით, აურიეთ და გაფილტრეთ. თუ ფილტრატი არ არის სრულყოფილად სუფთა (რაც იშვიათია), განმეორებით გაიმეორეთ განსაზღვრა უფრო დიდი რაოდენობით I და II Carrez-ის ხსნარებით, მაგალითად, 10 მლ.
გაზომეთ ხსნარის ოპტიკური ბრუნვა (როტაცია) 200 მმ მილში პოლარიმეტრით ან საქარიმეტრით.
5.3. 40% ეთანოლში ხსნადი ნივთიერებების ოპტიკური ბრუნვის (როტაციის) (P 'ან S') განსაზღვრა
აწონეთ 5 გ ნიმუში 1 მგ-მდე სიზუსტით, მოათავსეთ 100 მლ გრადირებულ (დანაყოფებიან) კოლბაში და დაამატეთ დაახლოებით 80 მლ ეთანოლი (ამ თავის პუნქტი 3.5) (იხ. დაკვირვება ამ თავის პუნქტი 7.2-ში). გააჩერეთ კოლბა 1 საათის განმავლობაში ოთახის ტემპერატურაზე; ამ დროის განმავლობაში, ექვსჯერ ენერგიულად შეანჯღრიეთ ისე, რომ გამოსაკვლევი ნიმუში საფუძვლიანად შეერიოს ეთანოლს. ეთანოლი შეავსეთ საჭირო მოცულობამდე (ამ თავის პუნქტი 3.5), აურიეთ და გაფილტრეთ.
პიპეტით გადაიტანეთ 50 მლ ფილტრატი (შეესაბამება 2,5 გ ნიმუშს) 250 მლ ერლენმეიერის კოლბაში, დაამატეთ 2,1 მლ მარილმჟავა (ამ თავის პუნქტი 3.1) და ენერგიულად შეანჯღრიეთ. ერლენმეიერის კოლბაში მოათავსეთ უკუმაცივარი (რეფლუქსის კონდენსატორი) და ჩაუშვით მდუღარე წყლის აბაზანაში. ზუსტად 15 წუთის შემდეგ ამოიღეთ ერლენმეიერის კოლბა აბაზანიდან, გადაიტანეთ შემცველობა 100 მლ გრადირებულ (დანაყოფებიან) კოლბაში, ჩემორეცხეთ მცირე რაოდენობის ცივი წყლით და გააგრილეთ 20oC ტემპერატურამდე.
დააზუსტეთ Carrez-ის I (ამ თავის პუნქტი 3.3) და II (ამ თავის პუნქტი 3.4) ხსნარების გამოყენებით, შეავსეთ საჭირო მოცულობამდე წყლით, ურიეთ, გაფილტრეთ და გაზომეთ ოპტიკური ბრუნვა, როგორც ეს აღნიშნულია ამ თავის 5.2 პუნქტის მე-2 და მე-3 ქვეპუნქტებში.
6. შედეგების დაანგარიშება სახამებლის შემცველობა (%) გამოითვლება შემდეგნაირად:
6.1. პოლარიმეტრით გაზომვა
P = მთლიანი ოპტიკური ბრუნვა კუთხის გრადუსებში; P'= 40% (V/V) ეთანოლში ხსნადი ნივთიერებების კუთხის გრადუსში ოპტიკური ბრუნვა; [α]D20=სუფთა სახამებლის სპეციფიკური ოპტიკური ბრუნვა. ამ ფაქტორისთვის პირობითად მიღებული რიცხვითი მნიშვნელობები არის შემდეგი:
6.2. საქარიმეტრით გაზომვა
S = სრული ოპტიკური ბრუნვა საქარიმეტრის გრადუსებში; S' = ოპტიკური ბრუნვა 40% ეთანოლში ხსნადი ნივთიერებების საქარიმეტრის გრადუსებში; N = საქაროზის წონა (გ) 100 მლ წყალში, რომელსაც გააჩნია ოპტიკური ბრუნვა 100 საქარიმეტრის გრადუსით, როდესაც იზომება 200 მმ მილის (ტუბის) გამოყენებით; 16,29 გ საფრანგეთის საქარიმეტრიტებისთვის; 26,00 გ გერმანიის საქარიმეტრიებისთვის; 20,00 გ შერეული საქარიმეტრისთვის.
[α]D20 = სუფთა სახამებლის სპეციფიკური ოპტიკური ბრუნვა (იხ. ამ თავის პუნქტი 6.1)
6.3. განმეორებადობა იმავე ნიმუშზე ჩატარებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 0,4-ს აბსოლუტურ მნიშვნელობაში, 40%-ზე ნაკლები სახამებლის შემცველობისათვის, და 1%-ს შედარებით მნიშვნელობაში, სახამებლის შემცველობისთვის, რომელიც ტოლი ან მეტია 40%-ისა.
7. დაკვირვება 7.1. თუ ნიმუში შეიცავს 6%-ზე მეტ კარბონატს, რომელიც გამოანგარიშებულია კალციუმის კარბონატის მიხედვით, ისინი უნდა განადგურდეს განზავებული გოგირდმჟავას ზუსტად შესაბამისი რაოდენობის დამუშავებით, მთლიანი ოპტიკური ბრუნვის განსაზღვრამდე. 7.2. ლაქტოზას მაღალი შემცველობის მქონე პროდუქტების შემთხვევაში, როგორიცაა რძის შრატის ფხვნილი ან გაუცხიმოვნებული რძის ფხვნილი, 80 მლ ეთანოლის დამატების შემდეგ (ამ თავის პუნქტი 3.5), მოქმედებთ შემდეგნაირად: კოლბში დაამაგრეთ უკუმაცივარი (რეფლუქსის კონდენსატორი) და ჩაუშვით წყლის აბაზანაში 50oC ტემპერატურაზე 30 წუთის განმავლობაში. დატოვეთ გასაგრილებლად და გააგრძელეთ გამოკვლევა, როგორც ეს მითითებულია ამ თავის 5.3 პუნქტში. 7.3. როდესაც პოლარიმეტრიული მეთოდით ხდება სახამებლის შემცველობის განსაზღვრა, ქვემოთ განსაზღვრული ცხოველის საკვები მასალა თუ მნიშვნელოვანი რაოდენობითაა წარმოდგენილი ცხოველის საკვებში, როგორც ცნობილია, იწვევს ხელის შეშლას და შეიძლება მოგვცეს არასწორი შედეგები: ა) (შაქარი) ჭარხლის პროდუქტები, როგორიცაა (შაქარი) ჭარხლის რბილობი, (შაქარი) ჭარხლის ბადაგი, (შაქარი) ჭარხლის რბილობი – ბადაგი, (შაქარი) ჭარხლის ვინასა, (ჭარხლის) შაქარი; ბ) ციტრუსის რბილობი; გ) სელის თესლი; სელის ნაწნეხი (ექსპელერი); ექსტრაჰირებული სელი; დ) რაპსის თესლი; რაპსის ნაწნეხი (ექსპელერი); ექსტრაჰირებული რაპსის თესლი, რაპსის თესლის ჩენჩო; ე) მზესუმზირის თესლი; ექსტრაჰირებული მზესუმზირის თესლი; მზესუმზირის თესლი, ნაწილობრივ ქერქგაცლილი, ექსტრაჰირებული; ვ) კოპრას (ქოქოსის კაკლის გამომშრალი გული) ნაწნეხი (ექსპელერი); ექსტრაჰირებული კოპრა; ზ) კარტოფილის რბილობი; თ) დეჰიდრატირებული საფუარი; ი) ინულინით მდიდარი პროდუქტები (მაგ., ჩიპსები და იერუსალიმის არტიშოკის (ტოპინამბურის) მსხვილად ნაფქვავი); კ) ხიწიწი. თავი XIII ნედლი ნაცრის განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას გვაძლევს, განვსაზღვროთ ნედლი ნაცრის შემცველობა ცხოველის საკვებში. 2. გამოყენების პრინციპი ნიმუში ნაცრდება (იხსნება) 550o C-ზე; ნაშთი (ნარჩენი) იწონება. 3. რეაგენტები ამონიუმის ნიტრატი, ხსნარი 20% (წ/მ). 4. აპარატურა 4.1. ცხელი ფირფიტა (პლიტა). 4.2. ელექტრომუფელური ღუმელი თერმოსტატით. 4.3. თიგელი დანაცრებისათვის, რომელიც დამზადებულია სილიციუმის, ფაიფურის ან პლატინისგან, მართკუთხა (დაახლ. 60 × 40 × 25 მმ) ან წრიული ფორმით (დიამეტრი: 60-დან 75 მმ-მდე, სიმაღლე: 20-დან 40 მმ-მდე). 5. გამოყენების პროცედურა აწონეთ მგ-მდე სიზუსტით დაახლოებით 5 გ ნიმუში (2,5 იმ შემთხვევაში, თუ გაჯირჯვების (შეშუპების) ტენდენცია აქვთ) და დანაცრებისთვის მოათავსეთ თიგელში, რომელიც პირველად გაცხელდა 550oC ტემპერატურაზე, გაცივდა და აიწონა. განათავსეთ თიგელი ცხელ ფირფიტაზე (პლიტაზე) და თანდათანობით გაათბეთ, სანამ ნივთიერება არ დანახშირდება. დაანაცრეთ ამ თავის 5.1 ან 5.2 პუნქტებით განსაზღვრული პირობების შესაბამისად. 5.1. განათავსეთ თიგელი დაკალიბრებულ მუფელურ ღუმელში, რომელიც დაყენებულია 550oC ტემპერატურაზე. დააყოვნეთ ამ ტემპერატურაზე, სანამ არ მიიღება თეთრი, ღია ნაცრისფერი ან მოწითალო ნაცარი, რომელიც თავისუფალი უნდა იყოს ნახშირბადის ნაწილაკებისგან. მოათავსეთ თიგელი დესიკატორში (საშრობში), დატოვეთ გასაგრილებლად და დაუყოვნებლივ აწონეთ. 5.2. მოათავსეთ თიგელი დაკალიბრებულ მუფელურ ღუმელში, რომელიც დაყენებულია 550oC ტემპერატურაზე. დაანაცრეთ 3 საათის განმავლობაში. განათავსეთ თიგელი დესიკატორში (საშრობში), დატოვეთ გასაცივებლად და დაუყოვნებლივ აწონეთ. დაანაცრეთ კიდევ ერთხელ 30 წუთის განმავლობაში, რათა დარწმუნდეთ, რომ ნაცრის წონა უცვლელი რჩება (წონის დანაკარგი ორ თანმიმდევრულ წონას შორის უნდა იყოს 1 მგ-ზე ნაკლები ან ტოლი). 6. შედეგების დაანგარიშება გამოთვალეთ ნაშთის (ნარჩენის) წონა ტარის გამოკლებით. გამოხატეთ შედეგი, როგორც ნიმუშის პროცენტული მაჩვენებელი. 7. დაკვირვება 7.1. ძნელად დასანაცრებელი ნივთიერებების ნაცარი უნდა დაექვემდებაროს თავდაპირველ დანაცრებას სულ მცირე სამი საათის განმავლობაში, რომელსაც უნდა მოსდევდეს გაგრილება და შემდეგ უნდა დაემატოს მას რამდენიმე წვეთი ამონიუმის ნიტრატის 20%-იანი ხსნარი ან წყალი (ფრთხილად იყავით, რომ არ მოხდეს ნაცრის გაფანტვა (დისპერსია) ან მთლიან მასად შედედება). ღუმელში გაშრობის შემდეგ განაგრძეთ დანაცრება. ოპერაცია გაიმეორეთ საჭიროებისდა მიხედვით, სანამ დანაცრება არ დასრულდება. 7.2. ამ თავის 7.1 ქვეპუნქტში აღწერილ დამუშავებაზე რეზისტენტული ნივთიერებების შემთხვევაში იმოქმედეთ შემდეგნაირად: სამი საათის დანაცრების შემდეგ, ნაცარი მოათავსეთ თბილ წყალში და გაფილტრეთ პატარა, ნაცრისაგან თავისუფალი ფილტრის საშუალებით. ფილტრი და მისი შემცველობა დანაცრდება თავდაპირველ თიგელში. მოათავსეთ ფილტრატი გაგრილებულ თიგელში, ააორთქლეთ სანამ არ გაშრება, დაანაცრეთ და აწონეთ. 7.3. ზეთებისა და ცხიმების შემთხვევაში, ზუსტად აწონეთ 25-გრამიანი ნიმუში შესატყვისი ზომის თიგელში. დაანახშირეთ მასალის აალებით, ნაცრისაგან თავისუფალი ფილტრის ქაღალდის დახმარებით. წვის შემდეგ დაატენიანეთ რაც შეიძლება ნაკლები წყლის რაოდენობით. გააშრეთ და მოახდინეთ დანაცრება, როგორც ეს აღწერილია ამ თავის მე-5 პუნქტში. თავი XIV მარილმჟავაში უხსნადი ნაცრის განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას გვაძლევს განვსაზღვროთ მარილმჟავაში უხსნადი მინერალური ნივთიერებების შემცველობა ცხოველის საკვებში. ნიმუშის ბუნებიდან გამომდინარე, შეიძლება ორი მეთოდის გამოყენება. 1.1. მეთოდი A: გამოიყენება ცხოველის საკვების ორგანული მასალებისთვის და ცხოველის კომბინირებული საკვების უმეტესობისთვის. 1.2. მეთოდი B: გამოიყენება მინერალური ნაერთების, ნარევებისა და ცხოველის კომბინირებული საკვებისათვის, რომელშიც მარილმჟავაში უხსნადი ნივთიერებების შემცველობა (რომელიც განსაზღვრულია A მეთოდით) აღემატება 1% -ს.
2. გამოყენების პრინციპი 2.1. მეთოდი A: ნიმუში დანაცრდება, ნაცარი დუღდება მარილმჟავაში და ხსნადი ნაშთი (ნარჩენები) იფილტრება და იწონება. 2.2. მეთოდი B: ნიმუში მუშავდება მარილმჟავათი. ხსნარი იფილტრება, ხდება ნაშთის (ნარჩენების) დანაცრება და ამგვარად მიღებული ნაცარი დამუშავდება A მეთოდის შესაბამისად.
3. რეაგენტები 3.1. მარილმჟავა 3 მოლი/ლიტრი. 3.2. ტრიქლორძმარმჟავა, ხსნარი 20% -იანი ხსნარი (წ/მ.). 3.3. ტრიქლორძმარმჟავა, ხსნარი 1% (წ/მ).
4. აპარატურა 4.1. ცხელი ფირფიტა (პლიტა). 4.2. ელექტრომუფელური ღუმელი თერმოსტატით. 4.3. თიგელი დანაცრებისთვის, რომელიც დამზადებულია სილიციუმის, ფაიფურის ან პლატინისგან, მართკუთხა (დაახლ. 60 × 40 × 25 მმ) ან წრიული ფორმით (დიამეტრი: 60-დან 75 მმ-მდე, სიმაღლე: 20-დან 40 მმ-მდე).
5. გამოყენების პროცედურა
5.1. მეთოდი A ხდება ნიმუშის დანაცრება იმ მეთოდის გამოყენებით, რომელიც აღწერილია ნედლი ნაცრის განსაზღვრისთვის. ასევე შეიძლება გამოყენებულ იქნეს ამ გამოკვლევის შედეგად მიღებული ნაცარი.
ნაცარი მოათავსეთ 250-დან 400-მლ-იან მენზურაში 75 მლ მარილმჟავას გამოყენებით (ამ თავის პუნქტი 3.1). ნელ-ნელა მიიყვანეთ დუღილის მდომარეობამდე და ნაზად ადუღეთ 15 წუთი. გაფილტრეთ თბილი ხსნარი ნაცრისგან თავისუფალი (უნაცრო) ფილტრის ქაღალდით და ჩამორეცხეთ ნაშთი (ნარჩენები) თბილი წყლით, სანამ მჟავას რეაქცია არ გაქრება. ნაშთის (ნარჩენების) და ნაცრის შემცველი ფილტრი გააშრეთ აწონილ თიგელში არანაკლებ 550oC ტემპერატურაზე და არაუმეტეს 700oC ტემპერატურაზე. გააგრილეთ დესიკატორში (საშრობში) და აწონეთ.
5.2. მეთოდი B აწონეთ 5 გ ნიმუში მგ-მდე სიზუსტით და მოათავსეთ 250-დან 400-მლ-იან მენზურაში. დაამატეთ 25 მლ წყალი და 25 მლ მარილმჟავა (ამ თავის პუნქტი 3.1) თანმიმდევრულად, შემდეგ აურიეთ და დაელოდეთ შუშხუნის შეწყვეტას. დაამატეთ კიდევ 50 მლ მარილმჟავა (ამ თავის პუნქტი 3.1). დაელოდეთ ვიდრე გაზის გამოთავისუფლება არ შეჩერდება, შემდეგ მოათავსეთ მენზურა მდუღარე წყლის აბაზანაში და დატოვეთ იგი 30 წუთის განმავლობაში ან უფრო მეტხანს, საჭიროების შემთხვევაში, რათა მოხდეს არსებული სახამებლის საფუძვლიანი ჰიდროლიზი. სანამ თბილია გაფილტრეთ ნაცრისგან თავისუფალი (უნაცრო) ფილტრის საშუალებით და გაწმინდეთ ფილტრი 50 მლ თბილ წყალში (იხ. ამ თავის პუნქტი 7 – დაკვირვება). ნაშთის (ნარჩენების) შემცველი ფილტრი მოათავსეთ თიგელში დანაცრებისთვის, გააშრეთ და მოახდინეთ დანაცრება არანაკლებ 550oC და არაუმეტეს 700oC ტემპერატურაზე. მოათავსეთ ნაცარი 250-დან 400-მლ-იან მენზურაში 75 მლ მარილმჟავას გამოყენებით (ამ თავის პუნქტი 3.1); შემდეგ გაგრძელეთ, როგორც ეს აღწერილია ამ თავის 5.1 პუნქტის მეორე აბზაცში.
6. შედეგების დაანგარიშება გამოითვალეთ ნაშთის (ნარჩენების) წონა ტარის გამოკლებით. გამოხატეთ შედეგი, როგორც ნიმუშის პროცენტული მაჩვენებელი.
7. დაკვირვება თუ ირკვევა, რონ ფილტრაცია რთულია, გამოკვლევა დაიწყეთ ახლიდან, 50 მლ მარილმჟავას (ამ თავის პუნქტი 3.1) ჩანაცვლებით 50 მლ 20% ტრიქლორძმარმჟავათი (ამ თავის პუნქტი 3.2) და გარეცხეთ ფილტრი 1% ტრიქლოროძმარმჟავას (ამ თავის პუნქტი 3.3) თბილი ხსნარით. თავი XV კარბონატების განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ კარბონატების რაოდენობა გამოხატული, როგორც კალციუმის კარბონატი, უმეტეს ცხოველის საკვებში. თუმცა, ზოგიერთ შემთხვევებში (მაგალითად, რკინის კარბონატით) სპეციალური მეთოდი უნდა იქნეს გამოყენებული.
2. გამოყენების პრინციპი კარბონატები იშლება მარილმჟავაში; გამოთავისუფლებული ნახშიროჟანგი გროვდება გრადირებულ (დანაყოფებიან) მილში და მისი მოცულობა დარდება იმავე პირობებში გამოთავისუფლებულ, კალციუმის კარბონატის ცნობილ რაოდენობასთან.
3. რეაგენტები 3.1. მარილმჟავა, სიმკვრივე 1,10 გ/მლ. 3.2. კალციუმის კარბონატი. 3.3. გოგირდმჟავა, დაახლოებით 0,05 მოლი/ლიტრი, შეფერადებული წითელი მეთილით.
4. აპარატურა Scheibler-Dietrich-ის აპარატი (იხ. დიაგრამა) ან ეკვივალენტური აპარატი.
5. გამოყენების პროცედურა ნიმუშის კარბონატული შემცველობის მიხედვით აწონეთ ნიმუშის პორცია, როგორც ნაჩვენებია ქვემოთ: – 0,5 გ იმ პროდუქტებისთვის, რომლებიც შეიცავს 50%-დან 100% კარბონატებს, და რომელიც გამოხატულია როგორც კალციუმის კარბონატი, – 1 გ იმ პროდუქტებისთვის, რომლებიც შეიცავს კარბონატების 40%-დან 50%-მდე, და რომელიც გამოხატულია როგორც კალციუმის კარბონატი, – 2-დან 3 გ-მდე სხვა პროდუქტებისთვის.
ნიმუშის პორცია მოათავსეთ აპარატის სპეციალურ კოლბაში (ამ თავის პუნქტი 4), რომელზედაც დაფიქსირებულია 10 მლ მარილმჟავას შემცველობის მქონე (ამ თავის პუნქტი 3.1), არამსხვრევადი მასალისგან დამზადებული მცირე ზომის მილი, და კოლბა შეუერთეთ აპარატს. გადაატრიალეთ სამმხრივი ონკანი (three-way cock) (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 5) ისე, რომ მილი (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 1) დაუკავშირდეს გარედან. მობილური მილის (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 2) გამოყენებით, რომელიც ივსება ფერადი გოგირდმჟავით (ამ თავის პუნქტი 3.3) და უერთდება საზომ (დანაყოფებიან) მილს (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 1), სითხის დონე მიიყვანეთ ნულოვან ნიშნულამდე. გადაატრიალეთ ონკანი (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 5), რათა მილები შეუერთოთ (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 1) და (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 3) და შეამოწმოთ, რომ დონე ნულოვან ნიშნულზეა.
მარილმჟავა (ამ თავის პუნქტი 3.1) ნელა გაატარეთ ნიმუშის პორციაზე, თან დახარეთ კოლბა (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 4). გაათანაბრეთ წნევა მილის (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 2) დაბლა დაშვებით. შეანჯღრიეთ კოლბა (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 4) სანამ ნახშირორჟანგის გამოყოფა მთლიანად არ შეჩერდება.
მილებში სითხის იმავე დონეზე დაუბრუნებით აღადგინეთ წნევა (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 1) და (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 2). რამდენიმე წუთის შემდეგ, როდესაც გაზის მოცულობა გახდება მუდმივი, აიღეთ მონაცემები.
ჩაატარეთ საკონტროლო ტესტი იმავე პირობებში 0,5 გ კალციუმის კარბონატზე (ამ თავის პუნქტი 3.2).
6. შედეგების დაანგარიშება კარბონატების შემცველობა, გამოხატული როგორც კალციუმის კარბონატი, გამოითვლება შემდეგი ფორმულით:
სადაც: X =% (წ/წ) კარბონატები ნიმუშში, გამოხატული როგორც კალციუმის კარბონატი; V = ნიმუშის პორციის მიერ გამოთავისუფლებული CO2 მლ; V1 = 0,5 გ CaCO3-ისგან გამოთავისუფლებული CO2 მლ; m = ნიმუშის პორციის წონა, გრამებში.
7. დაკვირვება 7.1. როდესაც ნიმუშის პორციის წონა 2 გ-ზე მეტია, კოლბაში ჯერ მოათავსეთ 15 მლ გამოხდილი წყალი (ამ თავის დიაგრამაზე ნაჩვენები წერტილი 4) და აურიეთ ტესტის დაწყებამდე. საკონტროლო ტესტისთვის გამოიყენეთ იმავე რაოდენობის წყალი. 7.2. თუ გამოყენებულ აპარატს აქვს Scheibler-Dietrich-ის აპარატისგან განსხვავებული მოცულობა, ნიმუშიდან და საკონტროლო ნივთიერებისაგან აღებული პორციები და შედეგების დაანგარიშება შესაბამისად უნდა იყოს ადაპტირებული.
Scheibler-Dietrich-ის CO 2 განსაზღვრის აპარატი
თავი XVI ფოსფორის საერთო რაოდენობის განსაზღვრა – ფოტომეტრიული მეთოდი 1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ ფოსფორის საერთო შემცველობა ცხოველის საკვებში, განსაკუთრებით მიზანშეწონილია ფოსფორის დაბალი შემცველობის პროდუქტების გამოკვლევისთვის. გარკვეულ შემთხვევებში (ფოსფორით მდიდარი პროდუქტი) შეიძლება გამოყენებულ იქნეს გრავიმეტრიული მეთოდი.
2. გამოყენების პრინციპი ნიმუში მინერალიზდება, ან მშრალი წვით (ცხოველის ორგანული საკვების შემთხვევაში) ან მჟავათი, გადახარშვის გზით (მინერალური ნაერთებისა და ცხოველის თხევადი საკვების შემთხვევაში) და თავსდება მჟავას ხსნარში. ხსნარი მუშავდება მოლიბდოვანადატის რეაგენტით. ამგვარად, წარმოქმნილი ყვითელი ხსნარის ოპტიკური სიმკვრივე იზომება სპექტროფოტომეტრში 430 ნმ-ზე.
3. რეაგენტები 3.1. კალციუმის კარბონატი. 3.2. მარილმჟავა, ρ 20 = 1,10 გ/მლ (დაახლ. 6 მოლი/ლიტრი). 3.3. აზოტის მჟავა, ρ 20 = 1,045 გ/მლ. 3.4. აზოტის მჟავა, ρ 20 = 1,38-დან 1,42 გ/მლ. 3.5. გოგირდის მჟავა, ρ 20 = 1,84 გ/მლ. 3.6. მოლიბდოვანადატის რეაგენტი: აურიეთ 200 მლ ამონიუმის ჰეპტამოლიბდატის ხსნარი (ამ თავის პუნქტი 3.6.1), 200 მლ ამონიუმის მონოვადატის ხსნარი (ამ თავის პუნქტი 3.6.2) და 134 მლ აზოტის მჟავა (ამ თავის პუნქტი 3.4) 1 ლიტრ საზომ (დანაყოფებიან) კოლბაში. შეავსეთ მოცულობა წყლით. 3.6.1. ამონიუმის ჰეპტამოლიბდატის ხსნარი: ცხელ წყალში გახსენით 100 გ ამონიუმის ჰეპტამოლიბდატი (NH 4) 6Mo7 O24·4H2O. დაამატეთ 10 მლ ამიაკი (სიმკვრივე 0,91 გ/მლ) და შეავსეთ 1 ლიტრამდე წყლით. 3.6.2. ამონიუმის მონოვადატის ხსნარი: გახსენით 2,35 გ ამონიუმის მონოვადატი NH 4 VO 3 400 მლ ცხელ წყალში. შეუჩერებლად ურიეთ, ნელა დაამატეთ 20 მლ გაზავებული აზოტის მჟავა (7 მლ HNO 3 (ამ თავის პუნქტი 3.4) + 13 მლ H 2 O და შეავსეთ 1 ლიტრამდე წყლით. 3.7. 1 მგ ფოსფორის 1 მლ სტანდარტული ხსნარი: წყალში გახსენით 4,387 გ კალიუმის დიჰიდროფოსფატი KH 2 PO 4. შეავსეთ 1 ლიტრამდე წყლით.
4. აპარატურა 4.1. სილიციუმის, ფაიფურის ან პლატინისგან დამზადებული თიგელი დანაცრებისათვის. 4.2. ელექტრომუფელური ღუმელი თერმოსტატით დაყენებული 550oC – ზე. 4.3. 250 მლ კელდალის კოლბა. 4.4. გრადირებული (დანაყოფებიანი) კოლბები და ზუსტი პიპეტები. 4.5. სპექტროფოტომეტრი. 4.6. დაახლოებით 16 მმ დიამეტრის მილები, გრადირებული (დანაყოფებიანი) სინჯარები 14,5 მმ დიამეტრის საცობები 14,5 მმ; მოცულობა: 25-დან 30 მლ-მდე.
5. გამოყენების პროცედურები
5.1. ხსნარის მომზადება ნიმუშის ბუნებიდან გამომდინარე, ხსნარი მოამზადეთ ისე, როგორც ეს მითითებულია ამ თავის 5.1.1 ან 5.1.2 ქვეპუნქტებში.
5.1.1. ჩვეულებრივი პროცედურა აწონეთ 1 გ ან მეტი ნიმუში 1 მგ სიზუსტით. ტესტის ნიმუში მოათავსეთ კელდალის კოლბაში, დაამატეთ 20 მლ გოგირდმჟავა (ამ თავის პუნქტი 3.5), შეანჯღრიეთ, რომ ნივთიერება მთლიანად გაიჟღენთოს მჟავით და რომ თავიდან აიცილოთ მისი კოლბის გვერდებზე მიკვრა, გაათბეთ და გააჩერეთ დუღილის წერტილზე 10 წუთის განმავლობაში. დატოვეთ ოდნავ რომ გაგრილდეს, დაამატეთ 2 მლ აზოტის მჟავა (ამ თავის პუნქტი 3.4), ოდნავ შეათბეთ, დატოვეთ, რომ ოდნავ გაგარილდეს, დაამატეთ ოდნავ მეტი აზოტის მჟავა (ამ თავის პუნქტი 3.4) და დააბრუნეთ დუღილის წერტილამდე. გაიმეორეთ ეს პროცედურა, სანამ არ მიიღებთ უფერო ხსნარს. გააგრილეთ, დაამატეთ ცოტაოდენი წყალი, ჩამორეცხეთ სითხე 500 მლ გრადირებულ (დანაყოფებიან) კოლბაში, თან შეაჯანჯღარეთ კელდალის კოლბა ცხელი წყლით. დატოვეთ გასაგრილებლად, შეავსეთ მოცულობა წყლით, მოახდინეთ ჰომოგენიზება და გაფილტრეთ.
5.1.2. ორგანული ნივთიერებების შემცველი და კალციუმის და მაგნიუმის დიჰიდროფოსფატებისგან თავისუფალი ნიმუშები აწონეთ დაახლოებით 2,5 გ ნიმუში 1 მგ-მდე სიზუსტემდე თიგელში . შეურიეთ გამოსაკვლევი ნიმუში, სანამ მთლიანად არ შეერევა 1 გ კალციუმის კარბონატს (ამ თავის პუნქტი 3.1). მოახდინეთ დანაცრება ღუმელში 550oC ტემპერატურაზე, სანამ არ მიიღება თეთრი ან ნაცრისფერი ნაცარი (ცოტაოდენ ნახშირს არ აქვს მნიშვნელობა). ნაცარი გადაიტანეთ 250 მლ მენზურაში. დაამატეთ 20 მლ წყალი და მარილმჟავა (ამ თავის პუნქტი 3.2) სანამ შუშხუნი არ შეჩერდება. დაამატეთ კიდევ 10 მლ მარილმჟავა (ამ თავის პუნქტი 3.2). მოათავსეთ მენზურა ქვიშის აბაზანაზე და ააორთქლეთ გაშრობამდე, რათა სილიციუმი შეიქმნას უხსნადი. ხელახლა გახსენით ნარჩენები 10 მლ აზოტმჟავაში (ამ თავის პუნქტი 3.3) და ადუღეთ ქვიშის აბაზანაზე ან ცხელ ფირფიტაზე 5 წუთის განმავლობაში, აორთქლების გარეშე, სანამ არ გაშრება. მოახდინეთ სითხის დეკანტირება 500 მლ გრადირებულ (დანაყოფებიან) კოლბაში, თან რამდენჯერმე ჩამორეცხეთ მენზურა ცხელი წყლით. დატოვეთ გასაგრილებლად, შეავსეთ მოცულობა წყლით, მოახდინეთ ჰომოგენიზება და გაფილტრეთ.
5.2. შეფერილობის შექმნა და ოპტიკური სიმკვრივის გაზომვა განაზავეთ ფილტრატის ალიკვოტის ნაწილი, რომელიც მოპოვებული იქნა ამ თავის 5.1.1 ან 5.1.2 პუნქტით, რათა მივიღოთ ფოსფორის კონცენტრაციის არაუმეტეს 40 მკგ/მლ. ამ ხსნარის 10 მლ მოათავსეთ სატესტო მილში (ამ თავის პუნქტი 4.6) და დაამატეთ 10 მლ მოლიბდოვანადატის რეაგენტი (ამ თავის პუნქტი 3.6). გააკეთეთ ჰომოგენიზაცია და გააჩერეთ სულ მცირე 10 წუთი 20°C ტემპერატურაზე. გაზომეთ ოპტიკური სიმკვრივე სპექტროფოტომეტრში 430 ნმ-ზე, რომელიც მიღებული იქნა ხსნარის 10 მლ მოლიბდოვანადატის რეაგენტის (ამ თავის პუნქტი 3.6) 10 მლ წყალში დამატებით.
5.3. საკალიბრო მრუდი სტანდარტული ხსნარიდან (ამ თავის პუნქტი 3.7) მოამზადეთ ხსნარები, რომლებიც შეიცავს 5, 10, 20, 30 და 40 მკგ ფოსფორს 1 მლ-ზე. აიღეთ თითოეული ამ ხსნარიდან 10 მლ და დაუმატეთ მას 10 მლ მოლიბდოვანანადის რეაგენტი (ამ თავის პუნქტი 3.6). გააკეთეთ ჰომოგენიზაცია და გააჩერეთ სულ მცირე 10 წუთი 20oC ტემპერატურაზე. გაზომეთ ოპტიკური სიმკვრივე, როგორც ეს მითითებულია ამ თავის 5.2-ე ქვეპუნქტში. მოახდინეთ საკალიბრო მრუდის მიკვლევა, ფოსფორის შესაბამის რაოდენობიდან ოპტიკური სიმკვრივის შედგენით. 0-დან 40 მკგ/მლ-მდე კონცენტრაციისთვის მრუდი იქნება წრფივი.
6. შედეგების დაანგარიშება ტესტის ნიმუშში განსაზღვრეთ ფოსფორის რაოდენობა საკალიბრო მრუდის გამოყენებით. გამოხატეთ შედეგი, როგორც ნიმუშის პროცენტული მაჩვენებელი.
განმეორებადობა
ერთი და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – მაღალი შედეგის 3%-ს, ფოსფორის 5% -ზე ნაკლები შემცველობისთვის; – 0,15%-ს აბსოლუტურ მნიშვნელობაში, ფოსფორის 5% ან მეტი შემცველობისთვის.
თავი XVII ქლორიდებიდან ქლორის განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ ქლორის რაოდენობა წყალში ხსნად ქლორიდებში, როგორც წესი ისინი გამოხატულია, როგორც ნატრიუმის ქლორიდი. იგი გამოიყენება ცხოველის ყველა საკვებისთვის.
2. გამოყენების პრინციპი ქლორიდები იხსნება წყალში. თუ პროდუქტი შეიცავს ორგანულ ნივთიერებებს, ხდება მისი გაწმენდა. ხსნარს ოდნავ ამჟავებენ აზოტის მჟავით და ქლორიდები ილექება ვერცხლის ქლორიდის სახით, ვერცხლის ნიტრატის ხსნარის გამოყენებით. ვერცხლის ნიტრატის ზედმეტობისას ხდება მისი ტიტრაცია ამონიუმის ციანსულფანიდის ხსნარით, Volhard-ის მეთოდის მიხედვით.
3. რეაგენტები 3.1. ამონიუმის ციანსულფანიდი (თიოციანატის) ხსნარი 0,1 მოლი/ლიტრი. 3.2. ვერცხლის ნიტრატის ხსნარი 0,1 მოლი/ლიტრი. 3.3. ამონიუმის რკინის სულფატის (NH4)Fe(SO4)2 გაჯერებული ხსნარი. 3.4. აზოტმჟავა, სიმკვრივე: 1,38 გ/მლ. 3.5. დიეთილის ეთერი. 3.6. აცეტონი. 3.7. Carrez ხსნარი I: წყალში გახსენით 21,9 გ თუთია აცეტატი, Zn (CH 3 COO) 2 · 2H 2 O და 3 გ დაკრისტალებული ძმარმჟავა. შეავსეთ 100 მლ-მდე წყლით. 3.8. Carrez ხსნარი II: წყალში გახსენით 10,6 გ კალიუმის ფეროციანიდი K4Fe(CN)6·3H2O. შეავსეთ 100 მლ-მდე წყლით. 3.9. აქტიური ნახშირბადი, ქლორიდებისაგან თავისუფალი და რომ არ იწოვდეს მათ.
4. აპარატურა მიქსერი (ტუმბლერი): დაახლოებით 35-დან 40-მდე წრებრუნვა წუთში.
5. გამოყენების პროცედურა
5.1. ხსნარის მომზადება ნიმუშის ბუნებიდან გამომდინარე, მოამზადეთ ხსნარი ისე, როგორც ეს ნაჩვენებია ამ თავის 5.1.1, 5.1.2 ან 5.1.3 ქვეპუნქტებში.
ამავდროულად ჩაატარეთ ბრმა (საკონტროლო) ტესტი გამოსაკვლევის ნიმუშის გარეშე.
5.1.1. ნიმუშები, რომლებიც არ შეიცავენ ორგანულ ნივთიერებებს აწონეთ მგ-მდე სიზუსტით ნიმუში არაუმეტეს 10 გ-ისა და რომელიც შეიცავს არაუმეტეს 3 გ ქლორს, ქლორიდების სახით. 400 მლ წყალთან ერთად მოათავსეთ 500 მლ მოცულობის კოლბაში, დაახლოებით 20oC ტემპერატურაზე. ურიეთ 30 წუთი შემრევ აპარატში (ტუმბოში), მიიყვანეთ მოცულობამდე, მოახდინეთ ჰომოგენიზაცია და გაფილტრეთ.
5.1.2. ორგანული ნივთიერებების შემცველი ნიმუშები, გარდა ამ თავის მე-5.1.3 ქვეპუნქტში ჩამოთვლილი პროდუქტებისა აწონეთ დაახლოებით 5 გ ნიმუში მგ-მდე სიზუსტით და მოათავსეთ 1 გ აქტიური ნახშირბადით 500 მლ მოცულობის კოლბაში. დაამატეთ 400 მლ წყალი დაახლოებით 20oC ტემპერატურაზე და 5 მლ Carrez-ის ხსნარი I (ამ თავის პუნქტი 3.7), ურიეთ 30 წამი, შემდეგ დაამატეთ 5 მლ Carrez II ხსნარი (ამ თავის პუნქტი 3.8). ურიეთ 30 წუთის განმავლობაში შემრევ აპარატში (ტუმბლერში), მიიყვანეთ მოცულობამდე, მოახდინეთ ჰომოგენიზაცია და გაფილტრეთ.
5.1.3. თერმულად დამუშავებული ცხოველის საკვები, სელის ნაწნეხი(ექსპელერი) და ფქვილი, სელის ფქვილით მდიდარი პროდუქტები და ლორწოვანი გარსით ან კოლოიდური ნივთიერებებით მდიდარი სხვა პროდუქტები (მაგალითად, დექსტრინირებული სახამებელი)
მოამზადეთ ხსნარი ისე, როგორც ეს აღწერილია ამ თავის 5.1.2-ე ქვეპუნქტში, მაგრამ არ გაფილტროთ. მოახდინეთ დეკანტირება (თუ საჭიროა ცენტრიფუგირება), ამოიღეთ 100 მლ სუპერნატანტი (ნალექზედა შრე) სითხე და გადაიტანეთ 200 მლ საზომ კოლბაში. აურიეთ აცეტონი (ამ თავის პუნქტი3.6) და მიიყვანეთ მოცულობამდე ამ გამხსნელით, მოახდინეთ ჰომოგენიზაცია და გაფილტრეთ.
5.2. ტიტრაცია პიპეტის გამოყენებით გადაიტანეთ ერლენმეიერის კოლბაში 25 მლ-დან 100 მლ-მდე ფილტრატი (სავარაუდო ქლორის შემცველობის მიხედვით), რომელიც მიღებულია ამ თავის 5.1.1, 5.1.2 ან 5.1.3.-ე ქვეპუნქტში აღწერილი მეთოდით. ალიკვოტის პორცია (გაყოფადი პორცია) არ უნდა შეიცავდეს 150 მგ ქლორზე მეტს (Cl). საჭიროების შემთხვევაში განაზავეთ არანაკლებ 50 მლ წყალთან ერთად, დაამატეთ 5 მლ აზოტის მჟავა (ამ თავის პუნქტი 3.4), ამონიუმის რკინის სულფატის (ამ თავის პუნქტი 3.3) 20 მლ გაჯერებული ხსნარი და ამონიუმის ციანსულფანიდი (თიოციანატის) ხსნარის ორი წვეთი (ამ თავის პუნქტი 3.1), გადატანილი ბიურეტის საშუალებით და ნულოვან ნიშნულამდე შევსებული. ბიურეტის გამოყენებით გადაიტანეთ ვერცხლის ნიტრატის ხსნარი (ამ თავის პუნქტი 3.2) ისე, რომ მიიღოთ 5 მლ ზედმეტობა. დაამატეთ 5 მლ დიეთილის ეთერი (ამ თავის პუნქტი 3.5) და ძლიერად შეანჯღრიეთ რათა მოახდინოთ ნალექის კოაგულაცია (შეწებება). მოახდინეთ ზედმეტი ვერცხლის ნიტრატის ტიტრაცია ამონიუმის ციანსულფანიდის (თიოციანატის) ხსნარით (ამ თავის პუნქტი 3.1), სანამ მოწითალო ყავისფერი ელფერი არ გასტანს ერთ წუთს.
6. შედეგების დაანგარიშება ქლორის (X) ოდენობა, გამოხატული ნატრიუმის ქლორიდის სახით, გამოითვლება შემდეგი ფორმულით:
სადაც:
V1 = 0,1 მოლი/ლ-ის დამატებული ვერცხლის ნიტრატის ხსნარის მლ-ია; V2 = 0,1 მოლი/ლ ამონიუმის ციანსულფანიდის (ამონიუმის თიოციანატის) ხსნარის მლ-ია, რომელიც გამოიყენება ტიტრაციისათვის; m = ნიმუშის წონა.
თუ ბრმა (საკონტროლო) ტესტი მიუთითებს, რომ მოხმარებულია ვერცხლის ნიტრატის ხსნარი 0,1 მოლი/ლ, გამოაკელით ეს მნიშვნელობა მოცულობას (V1 – V2).
7. დაკვირვება 7.1. ტიტრაცია შეიძლება განხორციელდეს პოტენციომეტრიით. 7.2. იმ პროდუქტების შემთხვევაში, რომლებიც ძალზე მდიდარია ზეთებით და ცხიმებით, პირველ რიგში, მოახდინეთ ცხიმის გამოცლა დიეთილის ეთერით ან პეტროლეუმის ეთერით. 7.3. თევზის ფქვილის შემთხვევაში, ტიტრაცია შეიძლება განხორციელდეს Mohr-ის მეთოდით.
დანართი №4
ცხოველის საკვებში ავტორიზებული დანამატების შემცველობის კონტროლისათვის გამოყენებული გამოკვლევის მეთოდები
თავი I A ვიტამინის განსაზღვრა
1. მიზანი და გამოყენების სფერო ამ მეთოდით შესაძლებელია განვსაზღვროთ A ვიტამის შემცველობა ცხოველის საკვებსა და პრემიქსებში. ვიტამინი A შეიცავს ყველა ტრანს-რეტინილის ალკოჰოლს და მის ცის-იზომერებს, რომლის განსაზღვრაც ხდება ამ მეთოდით. A ვიტამინის შემცველობა გამოხატულია საერთაშორისო ერთეულებში (IU) კგ-ზე. ერთი IU შეესაბამება 0,300 მკგ ყველა – ტრანს-ვიტამინის ალკოჰოლის, ან 0,344 მკგ ყველა-ტრანს-ვიტამინის აცეტატის, ან 0,550 მკგ ყველა-ტრანს-ვიტამინის პალმიტატის აქტივობას.
რაოდენობრივი შეფასების ზღვარი არის 2 000 IU ვიტამინი A/კგ.
2. გამოყენების პრინციპები ნიმუში ჰიდროლიზდება ეთანოლის კალიუმის ჰიდროქსიდის ხსნარით და A ვიტამინის ექსტრაჰირება ხდება პეტროლეუმის ეთერში. გამხსნელი ექსტრაჰირდება აორთქლების შედეგად და ნაშთი (ნარჩენები) იხსნება მეთანოლში და, საჭიროების შემთხვევაში, ზავდება საჭირო კონცენტრაციამდე. A ვიტამინის შემცველობა განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (RP-HPLC) UV ან ფლუორესცენტული დეტექტორის გამოყენებით. ქრომატოგრაფიული პარამეტრები შეირჩევა ისე, რომ არ მოხდეს განცალკევება ყველა-ტრანს-ვიტამინ A ალკოჰოლსა და მის ცის-იზომერებს შორის.
3. რეაგენტები 3.1. ეთანოლი, σ = 96% 3.2. პეტროლეუმის ეთერი, დუღილის დიაპაზონი 40 o C-60 o C 3.3. მეთანოლი 3.4. კალიუმის ჰიდროქსიდის ხსნარი, c = 50 გ/100 მლ 3.5. ნატრიუმის ასკორბატის ხსნარი, c = 10 გ/100 მლ (იხ. ამ თავის პუნქტი 7.7 – დაკვირვება) 3.6. ნატრიუმის სულფიდი, Na 2 S · x H 2 O (x = 7-9) 3.6.1. ნატრიუმის სულფიდის ხსნარი, c = 0,5 მოლი/ლიტრი გლიცერილში, ß = 120 გ/ლ (x = 9) (იხილეთ ამ თავის პუნქტი 7.8 – დაკვირვება) 3.7. ფენოლფთალინის ხსნარი, c = 2 გ/100 მლ ეთანოლში (ამ თავის პუნქტი 3.1) 3.8. 2-პროპანოლი 3.9. მობილური ფაზა HPLC-სთვის: მეთანოლის (ამ თავის პუნქტი 3.3) და წყლის ნარევი, მაგ. 980+20(v + v). ზუსტი თანაფარდობა განისაზღვრება გამოყენებული სვეტის მახასიათებლებით. 3.10. აზოტი, ჟანგბადის გარეშე 3.11. ყველა-ტრანს-ვიტამინ A-ს აცეტატი, განსაკუთრებით სუფთა, სერტიფიცირებული აქტივობით, მაგ. 2,80 x 10 6 IU/გ 3.11.1. ყველა-ტრანს-ვიტამინ A-ს აცეტატის მჟავას ხსნარი: აწონეთ 0,1 მგ-ის სიზუსტით 50 მგ ვიტამინ A-ს აცეტატი (ამ თავის პუნქტი 3.11) 100 მლ გრადირებულ კოლბაში. გახსენით 2-პროპანოლში (ამ თავის პუნქტი 3.8) და შეავსეთ იგი იმავე გამხსნელით. ამ ხსნარის ნომინალური კონცენტრაცია არის 1 400 IU ვიტამინი A, მლ-ზე. ზუსტი შემცველობა უნდა განისაზღვროს ამ თავის 5.6.3.1-ე პუნქტის შესაბამისად. 3.12. ყველა-ტრანს-ვიტამინ A-ს პალმიტატი, განსაკუთრებით სუფთა, სერტიფიცირებული აქტივობით, მაგ. 1,80 x 10 6 IU/გ 3.12.1. ყველა-ტრანს-ვიტამინ A-ს პალმიტატის საწყისი ხსნარი: აწონეთ 0,1 მგ-ს სიზუსტით 80 მგ ვიტამინ A-ს პალმიტატი (ამ თავის პუნქტი 3.12) 100 მლ გრადირებულ კოლბაში. გახსენით 2-პროპანოლში (ამ თავის პუნქტი 3.8) და შეავსეთ იგი იმავე გამხსნელით. ამ ხსნარის ნომინალური კონცენტრაცია არის 1 400 IU ვიტამინი A, მლ-ზე. ზუსტი შემცველობა უნდა განისაზღვროს ამ თავის 5.6.3.2-ე პუნქტის შესაბამისად. 3.13. 2,6-დი-ტერტ-ბუტილ-4-მეთილფენოლი (BHT) (იხ. ამ თავის პუნქტი 7.5 – დაკვირვება)
4. აპარატურა 4.1. ვაკუუმის როტორული (როტაციული) ამაორთქლებელი. 4.2. ქარვის მინის ჭურჭელი. 4.2.1. ბრტყელძირიანი ან კონუსური კოლბები, 500 მლ, გლუვზედაპირიანი მინის ბუდით 4.2.2. გრადირებული კოლბები გლუვზედაპირიანი მინის საცობებით, ვიწროყელიანი, 10, 25, 100 და 500 მლ. 4.2.3. კონუსური გაყოფი ძაბრი, 1 000 მლ, გლუვზედაპირიანი მინის საცობებით. 4.2.4. მსხლის ფორმის კოლბები, 250 მლ, გლუვზედაპირიანი მინის ბუდით 4.3. Allihn კონდენსატორი, გარსაცმის სიგრძე (jacket length) 300 მმ, გლუვზედაპირიანი მინის შეერთებით, გაზსადენის ადაპტორით. 4.4. ნაკეცებიანი (დაგოფრილი) ქაღალდის ფილტრები ფაზის გამოყოფისთვის, დიამეტრი 185 მმ (მაგ. Schleicher & Schuell 597 HY 1/2) 4.5. HPLC მოწყობილობა ინჯექტირების სისტემით 4.5.1. თხევადი ქრომატოგრაფიული სვეტი, 250 მმ x 4 მმ, C18, 5 ან 10 მკმ შეფუთვა, ან ეკვივალენტი (შესრულების კრიტერიუმი: მხოლოდ ერთი პიკი ყველა რეტინოლის იზომერისთვის HPLC პირობებში) 4.5.2. UV ან ფლუორესცენტის დეტექტორი, ცვალებადი ტალღის სიგრძის რეგულირებით 4.6. სპექტროფოტომეტრი 10 მმ კვარცის უჯრედებით 4.7. წყლის აბაზანა მაგნიტური სათქვეფელა 4.8. ექსტრაჰირების აპარატი (იხ. ამ თავის სურათი N1), რომელიც შედგება: 4.8.1. 1 ლ მოცულობის მინის ცილინდრისგან, რომელსაც აქვს გლუვი მინის ყელი და საცობი 4.8.2. გლუვი მინის ჩამსმელი, რომელსაც აქვს გვერდითი ტოტის მილი და სარეგულირებელი მილი, რომელიც ცენტრში გადის. სარეგულირებელი მილს უნდა ჰქონდეს U ფორმის ქვედა დაბოლოება და ფარსუნკა მოპირდაპირე მხარის ბოლოში, იმისთვის, რომ ცილინდრში არსებული სითხის ზედა ფენა გადატანილ იქნეს გამყოფ ძაბრში.
5. გამოყენების პროცედურა შენიშვნა: ვიტამინი A მგრძნობიარეა (UV-ულტრაიისფერი) სხივისა და დაჟანგვის მიმართ. ყველა ოპერაცია უნდა ჩატარდეს სინათლის (ქარვის მინის ჭურჭლის, ან ალუმინის ფოლგისგან დაცული მინის ჭურჭლის გამოყენებით) და ჟანგბადის (აზოტის მიწოდებით, კვებით) არარსებობის შემთხვევაში. ექსტრაჰირების დროს ჰაერი, რომელიც სითხეზე მაღლაა, უნდა შეიცვალოს აზოტით (საცობის დროდადრო მოშვებით, თავიდან აიცილეთ ზედმეტი წნევის დაგროვება).
5.1. ნიმუშის დამზადება დააქუცმაცეთ ნიმუში ისე, რომ მან გაიაროს 1 მმ-იანი ხვრელების მქონე საცერი, ყურადღება მიაქციეთ რომ არ დაუშვათ სითბოს გამოყოფა. დაფქვა უნდა განხორციელდეს უშუალოდ აწონვამდე და საპონიფიკაციამდე, წინააღმდეგ შემთხვევაში შეიძლება წარმოიშვას A ვიტამინის დანაკარგები.
5.2. საპონიფიკაცია A ვიტამინის შემცველობიდან გამომდინარე აწონეთ, 1 მგ-მდე სიზუსტით, 2 გრამიდან 25 გ-დე ნიმუში 500 მლ ბრტყელძირიან ან კონუსურ კოლბაში (ამ თავის პუნქტი 4.2.1). თანმიმდევრულად, წრიული ბრუნით, ჯერ დაამატეთ 130 მლ ეთანოლი (ამ თავის პუნქტი 3.1), დაახლოებით 100 მგ BHT (ამ თავის პუნქტი 3.13), 2 მლ ნატრიუმის ასკორბატის ხსნარი (ამ თავის პუნქტი 3.5) და შემდეგ, 2 მლ ნატრიუმის სულფიდის ხსნარი (ამ თავის პუნქტი 3.6). კოლბზე დაამონტაჟეთ კონდენსატორი (ამ თავის პუნქტი 4.3) და ჩაუშვით კოლბა წყლის აბაზანაში მაგნიტური სათქვეფელა(ამ თავის პუნქტი 4.7). გააცხელეთ ადუღებამდე და გააჩერეთ რეფლუქსისთვის 5 წუთით. შემდეგ დაამატეთ 25 მლ კალიუმის ჰიდროქსიდის ხსნარი (ამ თავის პუნქტი 3.4) კონდენსატორის საშუალებით (ამ თავის პუნქტი 4.3) და გააჩერეთ რეფლუქსისთვის კიდევ 25 წთ შერევის რეჟიმში, აზოტის ნელი ნაკადის ქვეშ. შემდეგ ჩამორეცხეთ კონდენსატორი დაახლოებით 20 მლ წყლით და გააგრილეთ კოლბის შემცველობა ოთახის ტემპერატურაზე.
5.3. ექსტრაჰირება დეკანსაციის (ფილტრაციის) გზით, საპონიფიკაციის ხსნარი 250 მლ 1 000 მლ-მდე წყლის საერთო მოცულობის რაოდენობრივი ჩამობანით გადაიტანეთ გამყოფ ძაბრში (ამ თავის პუნქტი 4.2.3) ან ექსტრაჰირების აპარატში (ამ თავის პუნქტი 4.8). ჩამორეცხეთ საპონიფიკაციის კოლბა თანმიმდევრულად, ჯერ 25 მლ ეთანოლით (ამ თავის პუნქტი 3.1) და შემდეგ, 100 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და ჩამოსარეცხი სითხე გადაიტანეთ გამყოფ ძაბრში ან ექსტრაჰირების აპარატში. წყლისა და ეთანოლის პროპორცია კომბინირებულ ხსნარებში უნდა იყოს დაახლოებით 2:1. ენერგიულად შეანჯღრიეთ 2 წთ და მიეცით საშუალება დაილექოს 2 წუთის განმავლობაში.
5.3.1. ექსტრაჰირება გამყოფი ძაბრის გამოყენებით (ამ თავის პუნქტი 4. 2. 3)
ფენების გამოყოფის შემდეგ (იხ. ამ თავის პუნქტი 7.3 – დაკვირვება) პეტროლეუმის ეთერის ფენა გადაიტანეთ სხვა გამყოფ ძაბრში (ამ თავის პუნქტი 4.2.3). გაიმეორეთ ეს ექსტრაჰირება ორჯერ, 100 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და ორჯერ, 50 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2).
გარეცხეთ კომბინირებული ექსტრაქტები გამყოფ ძაბრში ორჯერ 100 მლ წყლის პორციით, ნაზი ტრიალით (ემულსიების წარმოქმნის თავიდან ასაცილებლად) და შემდეგ განმეორებითი შეჯანჯღარებით კიდევ 100 მლ წყლის პორციით, სანამ ფენოლფთალინის ხსნარის დამატებაზე წყალი არ გახდება უფერო (ამ თავის პუნქტი 3.7) (როგორც წესი, ოთხჯერ გარეცხვა საკმარისია). გარეცხილი ექსტრაქტი გაფილტრეთ მშრალი ნაკეცებიანი (გოფრირებული) ფილტრის საშუალებით, ფაზის გამოსაყოფად (ამ თავის პუნქტი 4.4), რათა მოაცილოთ დარჩენილი წყალი (suspended water) 500 მლ გრადირებულ კოლბაში (ამ თავის პუნქტი 4.2.2). 50 მლ პეტროლეუმის ეთერით ჩამორეცხეთ გამყოფი ძაბრი და ფილტრი (ამ თავის პუნქტი 3.2), შეავსეთ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და კარგად აურიეთ.
5.3.2. ექსტრაჰირება, ექსტრაჰირების აპარატით (ამ თავის პუნქტი 4. 8) როდესაც ფენები გამოიყოფა (იხილეთ ამ თავის პუნქტი 7.3 – დაკვირვება), მინის ცილინდრის საცობი შეცვალეთ (ამ თავის პუნქტი 4.8.1) გლუვი მინის ჩამსმელით (ამ თავის პუნქტი 4.8.2) და მოათავსეთ რეგულირებადი მილის U ფორმის ქვედა ბოლო ისე, რომ იგი ინტერფეისის (ზედაპირის) დონეზე ოდნავ ზემოთ იყოს. აზოტის ხაზის მხრიდან, გვერდითი რიჩაგის წნევის გამოყენებით, ზედა პეტროლეუმის ფენა გადაიტანეთ 1 000 მლ გამყოფ ძაბრში (ამ თავის პუნქტი 4.2.3). 100 მლ პეტროლეუმის ეთერი დაუმატეთ მინის ცილინდრს, საცობს (ამ თავის პუნქტი 3.2) და კარგად შეანჯღრიეთ. მიეცით ფენებს გამოყოფის საშუალება და ზედა ფენა გადაიტანეთ გამყოფ ძაბრში, როგორც ადრე. გაიმეორეთ ექსტრაჰირების პროცედურა 100 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2), შემდეგ ორჯერ 50 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და დაუმატეთ პეტროლეუმის ეთერის ფენები გამყოფ ძაბრს.
გარეცხეთ კომბინირებული პეტროლეუმის ეთერის ექსტრაქტები, როგორც ამ თავის 5.3.1.-ე პუნქტშია აღწერილი და გააგრძელეთ ისე, როგორც იმავე პუნქტშია არის აღწერილია.
5.4. HPLC-ისთვის ნიმუშის ხსნარის მომზადება
პიპეტით გადაიტანეთ პეტროლეუმის ეთერის ხსნარის ალიქვოტის პორცია (ამ თავის პუნქტი 5.3.1 ან 5.3.2-დან) 250 მლ მსხლის ფორმის კოლბაში (ამ თავის პუნქტი 4.2.4). როტორულ ამაორთქლებელზე ააორთქლეთ გამხსნელი გამოშრობამდე შემცირებული წნევით (ამ თავის პუნქტი 4.1), აბაზანის ტემპერატურაზე არაუმეტეს 40oC-ისა. აღადგინე ატმოსფერული წნევა აზოტის დაშვებით (ამ თავის პუნქტი 3.10) და კოლბა ამოიღეთ როტორული ამაორთქლებელიდან.
დარჩენილი გამხსნელი ამოიღეთ აზოტის ნაკადის საშუალებით (ამ თავის პუნქტი 3.10) და ნაშთი (ნარჩენები) დაუყოვნებლად გახსენით მეთანოლის (ამ თავის პუნქტი 3.3) ცნობილ მოცულობაში (10-100 მლ) (A ვიტამინის კონცენტრაცია უნდა იყოს 5 IU/მლ-დან 30 IU/მლ-მდე).
5.5. განსაზღვრა HPLC-ით ვიტამინი A იყოფა C18 საპირისპირო ფაზის სვეტით (ამ თავის პუნქტი 4.5.1) და კონცენტრაცია იზომება UV დეტექტორის (325 ნმ) ან ფლუორესცენტული დეტექტორის საშუალებით (აღგზნებდობა: 325 ნმ, გამონაბოლქვი: 475 ნმ) (ამ თავის პუნქტი 4.5.2). ამ თავის 5.4-ე პუნქტში მიღებული მეთანოლური ხსნარის ალიქვოტის პორცია (მაგ. 20 მკლ) შეიყვანეთ, და მოახდინეთ განზავება მოძრავი ფაზით (ამ თავის პუნქტი 3.9). გამოთვალეთ ერთი და იმავე ნიმუშის ხსნარის რამდენიმე ინჯექტირების საშუალო პიკის სიმაღლე (ფართობი) და საკალიბრო ხსნარების რამდენიმე ინჯექტირების საშუალო პიკის სიმაღლეები (ფართობები) (ამ თავის პუნქტი 5.6.2).
HPLC -ისგამოყენების პირობები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან. თხევადი:
5.6. დაკალიბრება 5.6.1. სტანდარტული სამუშაო ხსნარების მომზადება პიპეტით გადაიტანეთ 20 მლ ვიტამინ A აცეტატის საწყისი ხსნარი (ამ თავის პუნქტი 3.11.1) ან 20 მლ ვიტამინ A პალმიტატის საწყისი ხსნარი (ამ თავის პუნქტი 3.12.1) 500 მლ ბრტყელძირიან ან კონუსურ კოლბაში (ამ თავის პუნქტი 4.2.1) და მოახდინეთ ჰიდროლიზება, როგორც ეს აღწერილია ამ თავის 5.2-ე პუნქტში, მაგრამ BHT დამატების გარეშე. ამის შემდეგ, პეტროლეუმის ეთერით მოახდინეთ ექსტრაჰირება (ამ თავის პუნქტი 3.2) ამ თავის 5.3-ე პუნქტის შესაბამისად, და შეავსეთ 500 მლ-მდე პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2). მოახდინეთ ამ ექსტრაქტის 100 მლ-ს აორთქლება როტატორული ამაორთქლებლით (იხ. ამ თავის პუნქტი 5.4) თითქმის გაშრობამდე, აიღეთ დარჩენილი გამხსნელი აზოტის ნაკადის საშუალებით (ამ თავის პუნქტი 3.10) და ნარჩენი ხელახლა გახსენით 10,0 მლ მეთანოლში (ამ თავის პუნქტი 3.3). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 560 IU ვიტამინ A-ს მლ-ზე. ზუსტი შემცველობა უნდა განისაზღვროს ამ თავის 5.6.3.3-ე პუნქტის შესაბამისად. სტანდარტული სამუშაო ხსნარი, გამოყენებამდე უნდა იყოს ახალი მომზადებული.
ამ სტანდარტული სამუშაო ხსნარის 2,0 მლ, გადაიტანეთ პიპეტით 20 მლ გრადირებულ კოლბაში, შეავსეთ მეთანოლით ნიშნულამდე (ამ თავის პუნქტი 3.3) და აურიეთ. ამ განზავებული სტანდარტული სამუშაო ხსნარის ნომინალური კონცენტრაცია შეადგენს 56 IU ვიტამინ A-ს მლ-ზე.
5.6.2. საკალიბრო ხსნარებისა და საკალიბრო მრუდის მომზადება
1,0, 2,0, 5,0 და 10,0 მლ განზავებული სტანდარტული სამუშაო ხსნარი გადაიტანეთ 20-მლ-იანი გრადირებული კოლბების სერიაში, შეავსეთ ნიშნულამდე მეთანოლით (ამ თავის პუნქტი 3.3) და აურიეთ. ამ ხსნარების ნომინალური კონცენტრაცია შეადგენს 2,8, 5,6, 14,0 და 28,0 IU ვიტამინ A-ს თითოეულ მლ-ზე.
რამდენჯერმე შეიყვანეთ 20 მკლ თითოეული საკალიბრო ხსნარი და განსაზღვრეთ საშუალო პიკის სიმაღლეები (ფართობები). საშუალო პიკის სიმაღლეების (ფართობების) გამოყენებით გამოსახეთ საკალიბრო მრუდი UV კონტროლის შედეგების გათვალისწინებით (ამ თავის პუნქტი 5.6.3.3).
5.6.3. სტანდარტული (ტიპური) ხსნარების UV სტანდარტიზაცია
5.6.3.1. ვიტამინი A-ს აცეტატის საწყისი ხსნარი
პიპეტით გადაიტანეთ 2,0 მლ ვიტამინ A აცეტატის საწყისი ხსნარი (ამ თავის პუნქტი 3.11.1) 50 მლ გრადირებულ კოლბაში (ამ თავის პუნქტი 4.2.2) და ნიშნულამდე შეავსეთ 2-პროპანოლით (ამ თავის პუნქტი 3.8). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 56 IU ვიტამინ A-ს მლ-ზე. 3,0 მლ ამ განზავებული ვიტამინი A აცეტატის ხსნარი პიპეტეტით გადაიტანეთ 25 მლ გრადირებულ კოლბაში და ნიშნულამდე შეავსეთ 2-პროპანოლით (ამ თავის პუნქტი 3.8). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 6,72 IU ვიტამინ A-ს მლ-ლზე. გაზომეთ ამ ხსნარის UV სპექტრი 2-პროპანოლის (ამ თავის პუნქტი 3.8) მიმართ სპექტროფოტომეტრში (ამ თავის პუნქტი 4.6) 300 ნმ-დან 400 ნმ-მდე. ექსტინციის მაქსიმუმი უნდა იყოს 325 ნმ-დან 327 ნმ-მდე.
A ვიტამინის შემცველობის გამოთვლა:
5.6.3.2. ვიტამინ A პალმიტატის საწყისი ხსნარი პიპეტით გადაიტანეთ 2,0 მლ ვიტამინ A პალმიტატის საწყისი ხსნარი (ამ თავის პუნქტი 3.12.1) 50 მლ გრადირებულ კოლბაში (ამ თავის პუნქტი 4.2.2) და შეავსეთ 2-პროპანოლით (ამ თავის პუნქტი 3.8). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 56 IU ვიტამინ A-ს მლ-ზე. 3,0 მლ ამ განზავებული ვიტამინი A პალმიტატის ხსნარი პიპეტით გადაიტანეთ 25 მლ გრადირებულ კოლბაში და შეავსეთ 2-პროპანოლით (ამ თავის პუნქტი 3.8). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 6,72 IU ვიტამინ A-ს მლ-ზე. გაზომეთ ამ ხსნარის UV სპექტრი 2-პროპანოლის მიმართ (ამ თავის პუნქტი 3.8) სპექტროფოტომეტრში (4.6) 300 ნმ-დან 400 ნმ-მდე. ექსტინციის მაქსიმუმი უნდა იყოს 325 ნმ-დან 327 ნმ-მდე.
A ვიტამინის შემცველობის გამოთვლა:
5.6.3.3. A ვიტამინის სტანდარტული სამუშაო ხსნარი
პიპეტით გადაიტანეთ 3,0 მლ განუზავებული ვიტამინ A-ს სტანდარტული სამუშაო ხსნარი, რომელიც მომზადებულია ამ თავის 5.6.1-ე პუნქტის შესაბამისად, 50-მლ-იან გრადირებულ კოლბაში (ამ თავის პუნქტი 4.2.2) და შეავსეთ 2-პროპანოლით ნიშნულამდე (ამ თავის პუნქტი 3.8). ამ ხსნარის 5,0 მლ პიპეტით გადაიტანეთ 25 მლ გრადირებულ კოლბაში და შეავსეთ 2-პროპანოლით (ამ თავის პუნქტი 3.8). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 6,72 IU ვიტამინ A-ს მლ-ზე.
გაზომეთ ამ ხსნარის ულტრაიისფერი სპექტრი 2 – პროპანოლთან მიმართებაში (ამ თავის პუნქტი 3.8) სპექტროფოტომეტრში (ამ თავის პუნქტი 4.6) 300 ნმ-დან 400 ნმ-მდე. ექსტინციის მაქსიმუმი უნდა იყოს 325 ნმ-დან 327 ნმ-მდე.
A ვიტამინის შემცველობის გამოთვლა:
6. შედეგების დაანგარიშება
ვიტამინ A-ს პიკის საშუალო სიმაღლიდან (ფართობიდან) განსაზღვეთ ნიმუშის ხსნარის კონცენტრაცია IU/მლ საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.6.2). IU/კგ ნიმუშის A ვიტამინის შემცველობა w, მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის A ვიტამინის კონცენტრაცია (ამ თავის პუნქტი 5.4) IU/მლ-ში; V 1 = ნიმუშის ხსნარის მოცულობა (ამ თავის პუნქტი 5.4) მლ-ში; V 2 = ამ თავის 5.4-ე პუნქტში აღებული ალიქვოტის მოცულობა მლ-ში; m = გამოსაკვლევი პორციის წონა გ-ში; 7. დაკვირვება 7.1. A ვიტამინის დაბალი კონცენტრაციის მქონე ნიმუშებისთვის შეიძლება სასარგებლო იყოს ორი საპონიფიკაციის-მუხტის (წონა: 25 გ) პეტროლეუმის ეთერის ექსტრაქტების გაერთიანება ერთ ხსნარში HPLC-ის განსაზღვრისთვის. 7.2. გამოკვლევისთვის აღებული სინჯის წონა არ უნდა შეიცავდეს 2 გ-ზე მეტ ცხიმს. 7.3. თუ ფაზის გამოყოფა არ მოხდება, დაამატეთ დაახლოებით 10 მლ ეთანოლი (ამ თავის პუნქტი 3.1) ემულსიის დასაშლელად. 7.4. თევზის ქონის (ზეთის) და სხვა სუფთა ცხიმების დროს საპონიფიკაციის პერიოდი უნდა გაგრძელდეს 45-60 წუთი. 7.5. BHT-ის ნაცვლად შესაძლებელია ჰიდროქინონის გამოყენება. 7.6. ნორმალური ფაზის სვეტის გამოყენებით შესაძლებელია რეტინოლის იზომერების გამოყოფა. მაგრამ ამ შემთხვევაში, ყველა ცის და ტრანს იზომერების პიკების სიმაღლეები (ფართობები) უნდა შეჯამდეს გამოთვლებისთვის. 7.7. ნატრიუმის ასკორბატის ხსნარის ნაცვლად შეიძლება გამოყენებულ იქნეს დაახლოებით 150 მგ ასკორბინის მჟავა. 7.8. ნატრიუმის სულფიდის ხსნარის ნაცვლად შეიძლება გამოყენებულ იქნეს დაახლოებით 50 მგ EDTA. 7.9. რძის შემცვლელებში A ვიტამინის ანალიზის შემთხვევაში განსაკუთრებული ყურადღება უნდა მიექცეს: – საპონიფიკაციის დროს (ამ თავის პუნქტი 5.2): ნიმუშში არსებული ცხიმის რაოდენობის გამო, შეიძლება საჭირო გახდეს კალიუმის ჰიდროქსიდის ხსნარის რაოდენობის გაზრდა (ამ თავის პუნქტი 3.4), – ექსტრაჰირებისას (ამ თავის პუნქტი 5.3): ემულსიების არსებობის გამო შეიძლება საჭირო გახდეს წყლის/ეთანოლის 2:1 თანაფარდობის ადაპტაცია.
იმის შესამოწმებლად, იძლევა თუ არა კვლევის გამოყენებული მეთოდი საიმედო შედეგებს ამ სპეციალურ მატრიცაზე (რძის შემცვლელი), დამატებით გამოსაკვლევ პორციაზე გამოყენებული უნდა იქნეს აღდგენითი ტესტი. თუ აღდგენის მაჩვენებელი 80%-ზე დაბალია, გამოკვლევის შედეგი უნდა გამოსწორდეს აღდგენის მიზნით. 8. განმეორებადობა იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 15%-ს უფრო მაღალ შედეგთან მიმართებაში.
9. ერთობლივი კვლევის შედეგები ([6])
L = ლაბორატორიების რაოდენობა; n = ერთეულის მნიშვნელობების რაოდენობა; S r = განმეორებადობის სტანდარტული გადახრა; S R = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; r = განმეორებადობა; R = რეპროდუქციულობა (შედეგიანობა); CVr = განმეორებადობის ვარიაციის კოეფიციენტი; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი.
თავი II E ვიტამინის განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღროთ E ვიტამინის შემცველობა ცხოველის საკვებსა და პრემიქსებში. E ვიტამინის შემცველობა გამოხატულია მგ DL-α – ტოკოფეროლის აცეტატით კგ-ზე. 1 მგ DL-α – ტოკოფეროლის აცეტატი შეესაბამება 0,91 მგ DL-α – ტოკოფეროლს (ვიტამინი E).
რაოდენობრივი განსაზღვრის ზღვარია 2 მგ ვიტამინი E/კგ. რაოდენობრივი შეფასების ეს ზღვარი მიიღწევა მხოლოდ ფლუორესცენტული დეტექტორით. UV დეტექტორით რაოდენობრივი შეფასების ლიმიტია 10 მგ/კგ.
2. გამოყენების პრინციპი ნიმუში ჰიდროლიზდება ეთანოლის კალიუმის ჰიდროქსიდის ხსნარით და ვიტამინ E ექტრაჰირდება პეტროლეუმის ეთერში. გამხსნელი სცილდება აორთქლებით და ნაშთი (ნარჩენები) იხსნება მეთანოლში და, საჭიროების შემთხვევაში, განზავდება საჭირო კონცენტრაციამდე. E ვიტამინის შემცველობა განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (RP-HPLC), ფლუორესცენტის ან UV (ულტრაიისფერი) დეტექტორის გამოყენებით.
3. რეაგენტები 3.1. ეთანოლი, σ = 96%. 3.2. პეტროლეუმის ეთერი, დუღილის დიაპაზონი 40oC-60oC 3.3. მეთანოლი. 3.4. კალიუმის ჰიდროქსიდის ხსნარი, c = 50 გ/100 მლ. 3.5. ნატრიუმის ასკორბატის ხსნარი, c = 10 გ/100 მლ (იხ. ამ თვაის პუნქტი 7.7 – დაკვირვება). 3.6. ნატრიუმის სულფიდი, Na 2 S · x H2O (x = 7-9). 3.6.1. ნატრიუმის სულფიდის ხსნარი, c = 0,5 მოლი/ლიტრი გლიცეროლში, β = 120 გ/ლ. (x = სთვის) 9) (იხ. ამ თავის პუნქტი 7.8 – დაკვირვება). 3.7. ფენოლფთალინის ხსნარი, c = 2 გ/100 მლ ეთანოლში (ამ თავის პუნქტი 3.1). 3.8. მობილური ფაზა HPLC-სთვის: მეთანოლის (ამ თავის პუნქტი 3.3) და წყლის ნარევი, მაგ. 980 + 20 (v + v). ზუსტი თანაფარდობა განისაზღვრება გამოყენებული სვეტის მახასიათებლების მიხედვით. 3.9. აზოტი, ჟანგბადისგან თავისუფალი. 3.10. DL-α – ტოკოფეროლის აცეტატი, განსაკუთრებით სუფთა, სერტიფიცირებული აქტივობით. 3.10.1. DL-α – ტოკოფეროლის აცეტატის საწყისი ხსნარი: აწონეთ 0,1 მგ სიზუსტით 0,1 მგ, 100 მგ DL-α – ტოკოფეროლის აცეტატი (ამ თავის პუნქტი 3.10) 100 მლ გრადირებულ კოლბაში. გახსენით ეთანოლში (ამ თავის პუნქტი 3.1) და ნიშნულამდე შეავსეთ იმავე გამხსნელით. აღნიშნული ხსნარის 1 მლ შეიცავს 1 მგ DL-α – ტოკოფეროლის აცეტატს. (UV კონტროლისთვის იხილეთ ამ თავის პუნქტი 5.6.1.3; სტაბილიზაციისთვის იხილეთ ამ თავის პუნქტი 7.4 – დაკვირვება). 3.11. DL-α – ტოკოფეროლი, განსაკუთრებით სუფთა, სერტიფიცირებული აქტივობით. 3.11.1. DL-α – ტოკოფეროლის საწყისი ხსნარი: აწონეთ 0,1 მგ სიზუსტით, 100 მგ DL-α – ტოკოფეროლი (ამ თავის პუნქტი 3.10) 100 მლ გრადირებულ კოლბაში. გახსენით ეთანოლში (ამ თავის პუნქტი 3.1) და ნიშნულამდე შეავსეთ იმავე გამხსნელით. აღნიშნული ხსნარის 1 მლ შეიცავს 1მგ DL-α-ტოკოფეროლს. (UV კონტროლისთვის იხილეთ ამ თავის პუნქტი 5.6.2.3; სტაბილიზაციისთვის იხილეთ ამ თავის პუნქტი 7.4 – დაკვირვება). 3.12. 2,6-დი-ტერტ-ბუტილ-4-მეთილფენოლი (BHT) (იხ. ამ თავის პუნქტი 7.5 – დაკვირვება).
4. აპარატურა 4.1. როტორული ამაორთლებელი, ვიდეო ჩამწერი მოწყობილობით. 4.2. ქარვის მინის ჭურჭელი. 4.2.1. ბრტყელძირიანი ან კონუსური კოლბები, 500 მლ, გლუვზედაპირიანი მინის ბუდით. 4.2.2. გრადირებული კოლბები გლუვზედაპირიანი მინის საცობით, ვიწროყელიანი, 10, 25, 100 და 500 მლ. 4.2.3. გამყოფი ძაბრები, კონუსური, 1 000 მლ, გლუვზედაპირიანი მინის საცობებით. 4.2.4. მსხლის ფორმის კოლბები, 250 მლ, გლუვზედაპირიანი მინის ბუდით. 4.3. Allihn კონდენსატორი, გარსაცმის სიგრძე 300 მმ, გლუვზედაპირიანი მინის შეერთებით, გაზსადენის (გაზის მიწოდების) ადაპტორით. 4.4. ნაკეცებიანი (გოფრირებული) ფილტრის ქაღალდი ფაზის გამოყოფისთვის, დიამეტრი 185 მმ (მაგ. Schleicher & Schuell 597 HY 1/2). 4.5. HPLC მოწყობილობა ინჯექტირების სისტემით. 4.5.1. თხევადი ქრომატოგრაფიული სვეტი, 250 მმ × 4 მმ, C 18, 5 ან 10 მკმ შეფუთვა, ან ეკვივალენტი. 4.5.2. ფლუორესცენტული ან ულტრაიისფერი დეტექტორი, ცვალებადი ტალღის სიგრძით. 4.6. სპექტროფოტომეტრი 10 მმ კვარცის უჯრედებით. 4.7. წყლის აბაზანა მაგნიტური სათქვეფელა. 4.8. ექსტრაჰირების აპარატი (იხ. ამ თავის სურათი N1), რომელიც შედგება: 4.8.1. 1 ლ მოცულობის მინის ცილინდრი, რომელსაც აქვს გლუვი მინის ყელი და საცობი. 4.8.2. გლუვზედაპირიანი მინის ჩამსმელი ნაწილი, რომელსაც აქვს გვერდითი ტოტის მილი და სარეგულირებელი მილი, გადის ცენტრში. სარეგულირებელ მილს უნდა ჰქონდეს U ფორმის ქვედა დაბოლოება და ფარსუნკა საპირისპირო ბოლოში, რათა ცილინდრში სითხის ზედა ფენა გადატანილი იქნეს გამყოფ ძაბრში.
5. გამოყენების პროცედურა შენიშვნა: E ვიტამინი მგრძნობიარეა (UV-ულტრაიისფერი) სინათლისა და დაჟანგვის მიმართ. ყველა ოპერაცია უნდა ჩატარდეს სინათლის (ქარვის მინის ჭურჭლის, ან ალუმინის ფოლგისგან დაცული მინის ჭურჭლის გამოყენებით) და ჟანგბადის (აზოტის მიწოდებით, კვებით) არარსებობის შემთხვევაში. ექსტრაჰირების დროს ჰაერი, რომელიც სითხეზე მაღლაა, უნდა შეიცვალოს აზოტით (საცობის დროდადრო მოშვებით, თავიდან აიცილეთ ზედმეტი წნევის დაგროვება).
5.1. ნიმუშის მომზადება დააქუცმაცეთ ნიმუში ისე, რომ მან გაიაროს 1 მმ-იანი ხრელების მქონე საცერი, ყურადღება მიაქციეთ, რომ არ მოხდეს სითბოს გამოყოფა. დაფქვა უნდა განხორციელდეს უშუალოდ აწონვამდე და საპონიფიკაციამდე, წინააღმდეგ შემთხვევაში შეიძლება მოხდეს E ვიტამინის დანაკარგები.
5.2. საპონიფიკაცია E ვიტამინის შემცველობიდან გამომდინარე აწონეთ, 0,01 მგ-მდე სიზუსტით, 2 გრამიდან 25 გ-მდე ნიმუში 500 მლ ბრტყელძირიან ან კონუსურ კოლბაში (ამ თავის პუნქტი 4.2.1). თანმიმდევრულად წრიული ბრუნებით, დაამატეთ 130 მლ ეთანოლი (ამ თავის პუნქტი 3.1), დაახლოებით 100 მგ BHT (ამ თავის პუნქტი 3.12), 2 მლ ნატრიუმის ასკორბატის ხსნარი (ამ თავის პუნქტი 3.5) და 2 მლ ნატრიუმის სულფიდის ხსნარი (ამ თავის პუნქტი 3.6). კოლბზე დაამონტაჟეთ კონდენსატორი (ამ თავის პუნქტი 4.3) და კოლბა მოატავსეთ წყლის აბაზანაში მაგნიტური სათქვეფელათი (ამ თავის პუნქტი 4.7). გააცხელეთ ადუღებამდე და გააჩერეთ რეფლუქსისთვის 5 წუთით. შემდეგ დაამატეთ 25 მლ კალიუმის ჰიდროქსიდის ხსნარი (ამ თავის პუნქტი 3.4) კონდენსატორის საშუალებით (ამ თავის პუნქტი 4.3) და გააჩერეთ რეფლუქსისთვის კიდევ 25 წთ შერევის რეჟიმში, აზოტის ნელი ნაკადის ქვეშ. შემდეგ ჩამორეცხეთ კონდენსატორი დაახლოებით 20 მლ წყლით და გააგრილეთ კოლბის შემცველობა ოთახის ტემპერატურაზე.
5.3. ექსტრაჰირება დეკანტაციით (ფილტრაციით) საპონიფიკაციის ხსნარი 250 მლ 1 000 მლ-მდე წყლის საერთო მოცულობის რაოდენობრივი ჩამობანით გადაიტანეთ გამყოფ ძაბრში (ამ თავის პუნქტი 4.2.3) ან ექსტრაჰირების აპარატში (ამ თავის პუნქტი 4.8). ჩამორეცხეთ საპონიფიკაციო კოლბა თანმიმდევრულად, ჯერ 25 მლ ეთანოლით (ამ თავის პუნქტი 3.1) და შემდეგ, 100 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და ჩამოსარეცხი სითხე გადაიტანეთ გამყოფ ძაბრში ან ექსტრაჰირების აპარატში. წყლისა და ეთანოლის პროპორცია კომბინირებულ ხსნარებში უნდა იყოს დაახლოებით 2:1. ენერგიულად შეანჯღრიეთ 2 წთ და დააცადეთ დაილექვა 2 წუთის განმავლობაში.
5.3.1. ექსტრაჰირება გამყოფი ძაბრის გამოყენებით (ამ თავის პუნქტი 4. 2. 3) ფენების გამოყოფის შემდეგ (იხ. ამ თავის პუნქტი 7.3 – დაკვირვება) პეტროლეუმის ეთერის ფენა გადაიტანეთ სხვა გამყოფ ძაბრში (ამ თავის პუნქტი 4.2.3). გაიმეორეთ ეს ექსრაჰირება ორჯერ, 100 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და ორჯერ, 50 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2).
გარეცხეთ კომბინირებული ექსტრაქტები გამყოფ ძაბრში ორჯერ 100 მლ წყლის პორციით ნაზი ტრიალით (ემულსიების წარმოქმნის თავიდან ასაცილებლად) და შემდეგ განმეორებითი შეჯანჯღარებით კიდევ 100 მლ წყლის პორციით, სანამ ფენოლფთალინის ხსნარის დამატებაზე წყალი არ გახდება უფერო (ამ თავის პუნქტი 3.7) (როგორც წესი, ოთხჯერ გარეცხვა საკმარისია). გარეცხილი ექსტრაქტი გაფილტრეთ მშრალი ნაკეციანი (გოფრირებული) ფილტრის საშუალებით, ფაზის გამოსაყოფად (ამ თავის პუნქტი 4.4), რათა მოაცილოთ დარჩენილი წყალი (suspended water) 500 მლ გრადირებულ კოლბაში (ამ თავის პუნქტი 4.2.2). 50 მლ პეტროლეუმის ეთერით ჩამორეცხეთ გამყოფი ძაბრი და ფილტრი (ამ თავის პუნქტი 3.2), შეავსეთ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და კარგად აურიეთ.
5.3.2. ექსტრაჰირება, ექსრაჰირების აპარატით (ამ თავის პუნქტი 4. 8) როდესაც ფენები გამოიყოფა (იხილეთ ამ თავის პუნქტი 7.3 – დაკვირვება), მინის ცილინდრის საცობი შეცვალეთ (ამ თავის პუნქტი 4.8.1) გლუვზედაპირიანი მინის ჩამსმელით (ამ თავის პუნქტი 4.8.2) და მოათავსეთ რეგულირებადი მილის U ფორმის ქვედა ბოლო ისე, რომ იგი ინტერფეისის (ზედაპირის) დონეზე ოდნავ ზემოთ იყოს. აზოტის ხაზი გვერდითი რიჩაგის წნევის გამოყენებით, პეტროლეუმის ზედა ფენა გადაიტანეთ 1 000 მლ გამყოფ ძაბრში (ამ თავის პუნქტი 4.2.3). 100 მლ პეტროლეუმის ეთერი დაუმატეთ მინის ცილინდრს, საცობს (ამ თავის პუნქტი 3.2) და კარგად შეანჯღრიეთ. მიეცით ფენებს გამოყოფის საშუალება და ზედა ფენა გადაიტანეთ გამყოფ ძაბრში, როგორც ადრე. გაიმეორეთ ექსტრაჰირების პროცედურა 100 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2), შემდეგ ორჯერ 50 მლ პეტროლეუმის ეთერით (ამ თავის პუნქტი 3.2) და დაამატეთ ნიშნულამდე პეტროლეუმის ეთერის ფენები გამყოფ ძაბრს. გარეცხეთ კომბინირებული პეტროლეუმის ეთერის ექსტრაქტები, როგორც ამ თავის 5.3.1-ე პუნქტშია აღწერილია და გააგრძელეთ ისე, როგორც იმავე პუნქტით არის აღწერილი. 5.4. HPLC-ისთვის ნიმუშის ხსნარის მომზადება პიპეტით გადაიტანეთ პეტროლეუმის ეთერის ხსნარის ალიქვოტის პორცია (ამ თავის პუნქტი 5.3.1 ან 5.3.2-დან) 250 მლ მსხლის ფორმის კოლბაში (ამ თავის პუნქტი 4.2.4). გამხსნელი გამოშრობამდე ააორთქლეთ როტორულ ამაორთქლებელზე შემცირებული წნევით (ამ თავის პუნქტი 4.1), აბაზანის ტემპერატურაზე არაუმეტეს 40oC-ისა. აღადგინეთ ატმოსფერული წნევა აზოტის დაშვებით (ამ თავის პუნქტი 3.10) და კოლბა ამოიღეთ როტორული ამაორთქლებელიდან. დარჩენილი გამხსნელი ამოიღეთ აზოტის ნაკადის საშუალებით (ამ თავის პუნქტი 3.9) და ნაშთი (ნარჩენები) დაუყოვნებლივ გახსენით მეთანოლის (ამ თავის პუნქტი 3.3) ცნობილ მოცულობაში (10-100 მლ) (A ვიტამინის კონცენტრაცია უნდა იყოს 5 IU/მლ-დან 30 IU/მლ-მდე).
5.5. განსაზღვრა HPLC-ით E ვიტამინი იყოფა C18 შებრუნებული ფაზის სვეტით (ამ თავის პუნქტი 4.5.1) და კონცენტრაცია იზომება UV დეტექტორის (292 ნმ) ან ფლუორესცენტული დეტექტორის საშუალებით (აღგზნებდობა: 295 ნმ, ემისია: 330 ნმ) (ამ თავის პუნქტი 4.5.2).
ამ თავის 5.4-ე პუნქტში მიღებული მეთანოლური ხსნარის ალიქვოტის პორცია (მაგ. 20 მკლ) შეიყვანეთ და მოახდინეთ ელუირება მობილური ფაზით (ამ თავის პუნქტი 3.8). გამოთვალეთ ერთი და იმავე ნიმუშის ხსნარის რამდენიმე ინჯექტირების საშუალო პიკის სიმაღლე (ფართობი) და საკალიბრო ხსნარების რამდენიმე ინჯექტირების საშუალო პიკის სიმაღლეები (ფართობები) (ამ თავის პუნქტი 5.6.2).
HPLC -ისგამოყენების პირობები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს მოგვცემს.
5.6. დაკალიბრება (DL-α – ტოკოფეროლის აცეტატი ან DL-α – ტოკოფეროლი)
5.6.1. D L – α – ტოკოფეროლის აცეტატის სტანდარტი
5.6.1.1. სტანდარტული სამუშაო ხსნარის მომზადება პიპეტით გადაიტანეთ 25 მლ DL-α – ტოკოფეროლის აცეტატის საწყისი ხსნარი (3.10.1) 500 მლ ბრტყელძირიან ან კონუსურ კოლბაში (4.2.1) და მოახდინეთ ჰიდროლიზი, როგორც ეს აღწერილია 5.2-ში. ამის შემდეგ მოახდინეთ ექსტრაჰირება პეტროლეუმის ეთერით (3.2) 5.3-ის შესაბამისად და შეავსეთ 500 მლ პეტროლეუმის ეთერით. აღნიშნული ექსტრაქტის 25 მლ-ი აორთქლეთ როტორულ ამაორთქლებელზე (იხ. 5.4) თითქმის გაშრობამდე, აიღეთ დარჩენილი გამხსნელი აზოტის ნაკადის საშუალებით (3.9) და ნარჩენები ხალეახლა გახსენით 25,0 მლ მეთანოლში (3.3). ამ ხსნარის ნომინალური კონცენტრაცია შეადგენს 45,5 მკგ DL-α-ტოკოფეროლი მლ-ზე, რაც ეკვივალენტურია 50 მკგ DL-α-ტოკოფეროლის აცეტატისა მლ-ზე. სტანდარტული სამუშაო ხსნარი, გამოყენებამდე უნდა იყოს ახალი მომზადებული.
5.6.1.2. საკალიბრო ხსნარების და საკალიბრო მრუდის მომზადება 1,0, 2,0, 4,0 და 10,0 მლ სტანდარტული სამუშაო ხსნარი გადაიტანეთ 20-მლ-იანი გრადირებული კოლბების სერიაში, შეავსეთ ნიშნულამდე მეთანოლით (3.3) და აურიეთ. ამ ხსნარების ნომინალური კონცენტრაცია არის 2,5, 5,0, 10,0 და 25,0 მკგ/მლ DL-α – ტოკოფეროლის აცეტატი, ანუ 2,28, 4,55, 9,10 მკგ/მლ და 22, 8 მკგ/მლ DL-α – ტოკოფეროლი.
რამდენჯერმე შეიყვანეთ 20 მკლ თითოეული საკალიბრო ხსნარი და განსაზღვრეთ საშუალო პიკის სიმაღლეები (ფართობები). საშუალო პიკის სიმაღლის (ფართობის) გამოყენებით, გამოსახეთ საკალიბრო მრუდი.
5.6.1.3. DL-α-ტოკოფეროლის აცეტატის საწყისი ხსნარის UV (ულტრაიისფერი)-ის სტანდარტიზაცია (3.10.1) განაზავდეთ 5,0 მლ DL-α – ტოკოფეროლის აცეტატის საწყისი ხსნარი (3.10.1) 25,0 მლ-მდე ეთანოლით და გავზომეთ ამ ხსნარის ულტრაიისფერი სპექტრი ეთანოლის (3.1) მიმართ სპექტროფოტომეტრში (4.6) 250 ნმ-დან 320 ნმ-მდე.
შეწოვის მაქსიმუმი უნდა იყოს 284 ნმ.
ამ განზავებისას მოპოვებული უნდა იქნეს შთანთქმის 0,84-დან 0,88-მდე მნიშვნელობა.
5.6.2 DL-α – ტოკოფეროლის სტანდარტი 5.6.2.1. სტანდარტული სამუშაო ხსნარის მომზადება პიპეტით გადაიტანეთ 2 მლ DL-α-ტოკოფეროლის საწყისი ხსნარი (ამ თავის პუნქტი 3.11.1) 50 მლ გრადირებულ კოლბაში, გახსენით მეთანოლში (ამ თავის პუნქტი 3.3) და შეავსეთ ნიშნულამდე მეთანოლით. ამ ხსნარის ნომინალური კონცენტრაცია არის 40 მკგ DL-α – ტოკოფეროლი მლ-ზე, და ეკვივალენტურია 44,0 მკგ DL-α – ტოკოფეროლის აცეტატისა მლ-ზე. სტანდარტული სამუშაო ხსნარი, გამოყენებამდე უნდა იყოს ახალი მომზადებული. 5.6.2.2. საკალიბრო ხსნარების და საკალიბრო მრუდის მომზადება 1,0, 2,0, 4,0 და 10,0 მლ სტანდარტული სამუშაო ხსნარი გადაიტანეთ 20-მლ-იანი გრადირებული კოლბების სერიაში, შეავსეთ ნიშნულამდე მეთანოლით (ამ თავის პუნქტი 3.3) და აურიეთ. ამ ხსნარების ნომინალური კონცენტრაცია არის 2,0, 4,0, 8,0 და 20,0 მკგ/მლ DL-α – ტოკოფეროლი, ანუ 2,20, 4,40, 8,79 მკგ/მლ და 22,0 მკგ/მლ DL-α – ტოკოფეროლის აცეტატი. რამდენჯერმე შეიყვანეთ 20 მკლ თითოეული საკალიბრო ხსნარი და განსაზღვრეთ საშუალო პიკის სიმაღლეები (ფართობები). საშუალო პიკის სიმაღლის (ფართობის) გამოყენებით, გამოსახეთ საკალიბრო მრუდი. 5.6.2.3. DL-α-ტოკოფეროლის საწყისი ხსნარის UV (ულტრაიისფერი)-ის სტანდარტიზაცია (ამ თავის პუნქტი 3.11.1) განაზავეთ 2,0 მლ DL-α-ტოკოფეროლის საწყისი ხსნარი (ამ თავის პუნქტი 3.11.1) 25,0 მლ-მდე ეთანოლით და გაზომეთ ამ ხსნარის ულტრაიისფერი სპექტრი ეთანოლის მიმართ (ამ თავის პუნქტი 3.1) სპექტროფოტომეტრში (ამ თავის პუნქტი 4.6) 250 ნმ-დან 320-მდე. აბსორბციის (შთანთქმის) მაქსიმუმი უნდა იყოს 292 ნმ.
ამ განზავებისას მოპოვებული უნდა იქნეს 0,6 ექსტინციის მნიშვნელობა.
6. შედეგების დაანგარიშება გამოსაკვლევი ხსნარის E ვიტამინის პიკების საშუალო სიმაღლიდან (ფართობიდან) განისაზღვრება ნიმუშის ხსნარის კონცენტრაცია მკგ/მლ-ში (გამოითვლება α-ტოკოფეროლის აცეტატი) საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.6.1.2 ან 5.6.2.2).
E ვიტამინის ნიმუშის შემცველობა w/მგ კგ-ში მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის E ვიტამინის კონცენტრაცია (როგორც α – ტოკოფეროლის აცეტატი) (ამ თავის პუნქტი 5.4) IU/მლ-ში; V 1 = ნიმუშის ხსნარის მოცულობა (ამ თავის პუნქტი 5.4) მლ-ში; V 2 = ამ თავის 5.4-ე პუნქტში აღებული ალიქვოტის მოცულობა მლ-ში; m = გამოსაკვლევი პორციის წონა გ-ში. 7. დაკვირვება 7.1. E ვიტამინის დაბალი კონცენტრაციის მქონე ნიმუშებისთვის შეიძლება სასარგებლო იყოს ორი საპონიფიკაციის-მუხტის (წონა: 25 გ) პეტროლეუმის ეთერის ექსტრაქტების გაერთიანება ერთ ხსნარში HPLC-ის განსაზღვრისთვის. 7.2. გამოკვლევისთვის აღებული ნიმუშის წონა არ უნდა შეიცავდეს 2 გ-ზე მეტ ცხიმს. 7.3. თუ ფაზის გამოყოფა არ მოხდება, დაამატეთ დაახლოებით 10 მლ ეთანოლი (ამ თავის პუნქტი 3.1) ემულსიის დასაშლელად. 7.4. DL-α-ტოკოფეროლის აცეტატის ან DL-α-ტოკოფეროლის ხსნარის სპექტროფოტომეტრიული გაზომვის შემდეგ, ამ თავის 5.6.1.3 ან 5.6.2.3 პუნქტის შესაბამისად, ხსნარს (ამ თავის პუნქტი 3.10.1 ან 3.10.2) დაუმატეთ დაახლოებით 10 მგ BHT (ამ თავის პუნქტი 3.12) და ხსნარი შეინახეთ მაცივარში (შენახვის ვადა არის მაქსიმუმ 4 კვირა). 7.5. ჰიდროქინონის გამოყენება შესაძლებელია BHT-ის ნაცვლად. 7.6. ნორმალური ფაზის სვეტის გამოყენებით შესაძლებელია α-, β-, γ – და δ – ტოკოფეროლის გამოყოფა. 7.7. ნატრიუმის ასკორბატის ხსნარის ნაცვლად შეიძლება გამოყენებულ იქნეს დაახლოებით 150 მგ ასკორბინის მჟავა. 7.8. ნატრიუმის სულფიდის ხსნარის ნაცვლად შეიძლება გამოყენებულ იქნეს დაახლოებით 50 მგ EDTA. 7.9. E ვიტამინის აცეტატი ძალიან სწრაფად ჰიდროლიზდება ტუტე პირობებში და ამიტომ ძალიან მგრძნობიარეა ჟანგვის მიმართ, განსაკუთრებით ისეთი მიკროელემენტების არსებობისას, როგორიცაა რკინა ან სპილენძი. პრემიქსებში 5 000 მგ/კგ-ზე მეტი შემცველობით E ვიტამინის განსაზღვრის შემთხვევაში შეიძლება გამოიწვიოს ვიტამინის დეგრადაცია. ამიტომ, დასადასტურებლად რეკომენდებულია HPLC მეთოდი, რომელიც მოიცავს ვიტამინ E-ს ფორმულაციის ფერმენტულ დამუშავებას, ტუტე საპონიფიკაციის ეტაპის გარეშე. 8. განმეორებადობა იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 15%-ს უფრო მაღალ შედეგთან მიმართებაში. 9. ერთობლივი კვლევის შედეგები (7)
L = ლაბორატორიების რაოდენობა; n = ერთეულის მნიშვნელობების რაოდენობა; S r = განმეორებადობის სტანდარტული გადახრა; S R = სტანდარტული გადახრა; r = განმეორებადობა; R =რეპროდუქციულობა (შედეგიანობა); CVr = განმეორებადობის ვარიაციის კოეფიციენტი; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი.
თავი III რკინის, სპილენძის, მანგანუმისა და თუთიის კვალი ელემენტების განსაზღვრა
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა ცხოველის საკვებში განვსაზღვროთ რკინის, სპილენძის, მანგანუმის და თუთიის კვალი ელემენტები. რაოდენობრივი შეფასების შეზღუდვებია: – რკინა (Fe): 20 მგ/კგ – სპილენძი (Cu): 10 მგ/კგ – მანგანუმი (Mn): 20 მგ/კგ – თუთია (Zn): 20 მგ/კგ.
2. გამოყენების პრინციპი ნიმუში თავსდება ხსნარში, მარილმჟავაში, ორგანული ნივთიერებების განადგურების შემდეგ, ასეთის არსებობის შემთხვევაში. რკინის, სპილენძის, მანგანუმის და თუთიის ელემენტები განისაზღვრება შესაბამისი განზავების შემდეგ, ატომური აბსორბციული სპექტრომეტრიით.
3. რეაგენტები შესავალი კომენტარები
რეაგენტების და გამოსაკვლევი ხსნარების მოსამზადებლად გამოიყენეთ ბორსილიკატის ჭიქაში ან კვარცში წყლის ორმაგი გამოხდით ან იონგაცვლილ ფისზე ორმაგი დამუშავებით მიღებული კათიონებისაგან თავისუფალი წყალი, რომელიც უნდა განისაზღვროს.
რეაგენტები შესაფერისი უნდა იყოს ანალიტიკური შეფასებისთვის. განსასაზღვრი ელემენტის არ არსებობა უნდა შემოწმდეს საკონტროლო ექსპერიმენტისას. საჭიროების შემთხვევაში, უნდა მოხდეს რეაგენტების დამატებითი გაწმენდა.
ქვემოთ აღწერილი სტანდარტული ხსნარების ნაცვლად, შესაძლებელია გამოყენებულ იქნეს კომერციული სტანდარტული ხსნარები, თუ ისინი გარანტირებულია და გამოყენებამდე იქნა შემოწმებული. 3.1. მარილმჟავა (d: 1,19 გ/მლ). 3.2. მარილმჟავა (6 მოლი/ლიტრი). 3.3. მარილმჟავა (0,5 მოლი/ლიტრი). 3.4. ფტორწყალბადმჟავა 38%-დან 40%-მდე (v/v), რომელსაც აქვს 1 მგ/ლიტრზე ნაკლები რკინის (Fe) შემცველობა და ნაშთი (ნარჩენი) 10 მგ-ზე ნაკლები (სულფატის სახით)/ლიტრზე აორთქლების შემდეგ. 3.5. გოგირდმჟავა (დ: 1,84 გ/მლ). 3.6. წყალბადის ზეჟანგი (დაახლოებით 100 ჟანგბადის მოცულობა (წონა 30%)). 3.7. რკინის სტანდარტული ხსნარი (1000 მკგ Fe/მლ), რომელიც მზადდება შემდეგნაირად ან კომერციულად (გასაყიდად) გამოსაყენებელი ეკვივალენტური ხსნარი: გახსენით 1 გ რკინის მავთული 200 მლ 6 მოლ/ლიტრ მარილმჟავაში (ამ თავის პუნქტი 3.2), დაამატეთ 16 მლ წყალბადის ზეჟანგი (ამ თავის პუნქტი 3.6) და წყლით შეავსეთ ერთ ლიტრამდე. 3.7.1. რკინის სტანდარტული სამუშაო ხსნარი (100 მკგ Fe/მლ), მზადდება სტანდარტული ხსნარის ერთი წილის (ამ თავის პუნქტი 3.7), 9 ნაწილ წყალთან განზავებით. 3.8. სპილენძის სტანდარტული ხსნარი (1000 მკგ Cu/მლ), რომელიც მზადდება შემდეგნაირად, ან კომერციულად (სავაჭროდ) ხელმისაწვდომი ეკვივალენტური ხსნარი: – 1 გ სპილენძის ფხვნილი გახსენით 25 მლ 6 მოლ/ლიტრ მარილმჟავაში (ამ თავის პუნქტი 3.2), დაამატეთ 5 მლ წყალბადის ზეჟანგი (ამ თავის პუნქტი 3.6) და წყლით შეავსეთ ერთ ლიტრამდე. 3.8.1. სპილენძის სტანდარტული სამუშაო ხსნარი (10 მკგ Cu/მლ), რომელიც მზადდება სტანდარტული ხსნარის 1 წილის (ამ თავის პუნქტი 3.8) 9 წილ წყალთან განზავებით და შემდეგ, მიღებული ხსნარის 1 წილის 9 წილ წყალთან განზავებით. 3.9. მანგანუმის სტანდარტული ხსნარი (1 000 მკგ Mn/მლ) მომზადებულია შემდეგნაირად, ან კომერციულად (სავაჭროდ) ხელმისაწვდომი ეკვივალენტური ხსნარი: – 1 გ მანგანუმი ფხვნილი გახსენით 25 მლ 6 მოლ/ლიტრ მარილმჟავაში (ამ თავის პუნქტი 3.2) და წყლით შეავსეთ ერთ ლიტრამდე. 3.9.1. მანგანუმის სტანდარტული სამუშაო ხსნარი (10 მკგ Mn/მლ), რომელიც მზადდება სტანდარტული ხსნარის 1 წილის (ამ თავის პუნქტი 3.9) 9 წილ წყალთან განზავებით და შემდეგ, მიღებული ხსნარის 1წილის 9 წილ წყალთან განზავებით. 3.10. თუთიის სტანდარტული ხსნარი (1 000 მკგ Zn/მლ) მომზადებულია შემდეგნაირად, ან კომერციულად (სავაჭროდ) ხელმისაწვდომი ეკვივალენტური ხსნარი: – ზოლის ან ფოთლის ფორმის თუთიის 1 გ გახსენით 25მლ-ში 6 მოლ/ლიტრ მარილმჟავაში (ამ თავის პუნქტი 3.2) და შეავსეთ ერთ ლიტრამდე წყლით. 3.10.1. თუთიის სტანდარტული სამუშაო ხსნარი (10 მკგ Zn/მლ), რომელიც მზადდება სტანდარტული ხსნარის 1 ნაწილის (ამ თავის პუნქტი 3.10) წყალთან 9 ნაწილად განზავებით და შემდეგ, მიღებული ხსნარის 1 ნაწილის, წყალთან 9 ნაწილად განზავებით. 3.11. ლანთანის ქლორიდის ხსნარი: გახსენით 12 გ ლანთანის ოქსიდი 150 მლ წყალში, დაუმატეთ 100 მლ 6 მოლი/ლიტრი მარილმჟავას (ამ თავის პუნქტი 3.2) და შეავსეთ ერთ ლიტრამდე წყლით.
4. აპარატურა 4.1. მუფელური ღუმელი ტემპერატურის რეგულირებით და სასურველია ჩამწერით. 4.2. მინის ჭურჭელი უნდა იყოს რეზისტენტული ბოროსილიკატის ტიპის და რეკომენდებულია იმ აპარატების გამოყენება, რომლებიც განკუთვნილია მხოლოდ კვალი ელემენტების განსაზღვრისთვის. 4.3. ატომური აბსორბაციის სპექტროფოტომეტრი, რომელიც აკმაყოფილებს მეთოდის მოთხოვნებს საჭირო დიაპაზონში მგრძნობელობისა და სიზუსტის გათვალისწინებით.
5. პროცედურა (8) 5.1. ორგანული ნივთიერების შემცველი ნიმუშები 5.1.1. დანაცრება და ხსნარის მომზადება გამოკვლევისთვის (9) 5.1.1.1. მოათავსეთ 5-დან 10 გრამამდე ნიმუში 0,2 მგ-მდე სიზუსტით, კვარცის ან პლატინის თიგელში (იხ. ამ თავის შენიშვნა „ბ“), გააშრეთ ღუმელში 105oC ტემპერატურაზე და შეიტანეთ თიგელი ცივ მუფელურ ღუმელში (ამ თავის პუნქტი 4.1). დახურეთ ღუმელი (იხ. ამ თავის შენიშვნა „გ“) და თანდათან აუწიეთ ტემპერატურას 450-დან 475°C-მდე დაახლოებით 90 წუთის განმავლობაში. შეინარჩუნეთ ეს ტემპერატურა 4-დან 16 საათამდე (მაგ. ღამით) ნახშირბადოვანი მასალის მოსაცილებლად და შემდეგ გახსენით ღუმელი და გააგრილეთ (იხ. ამ თავის შენიშვნა „დ“). დაატენიანეთ ნაცარი წყლით და გადაიტანეთ 250 მლ მენზურაში. გარეცხეთ თიგელში დაახლოებით 5 მლ მარილმჟავით (ამ თავის პუნქტი 3.1) და დაამატეთ ეს უკანასკნელი ნელა და ფრთხილად მენზურაზე (შეიძლება იყოს ძლიერი რეაქცია CO2-ის წარმოქმნის გამო). დაუმატეთ მარილმჟავას (ამ თავის პუნქტი 3.1) წვეთ-წვეთობითი არევით, სანამ ყველა შუშხუნი არ შეჩერდება. ააორთქლეთ გამოშრობამდე, დროდადრო ურიეთ მინის წკირით. შემდეგ 15 მლ 6 მოლი/ლიტრ მარილმჟავა (ამ თავის პუნქტი 3.2) დაუმატეთ ნაშთს (ნარჩენს), შემდეგ დაახლოებით 120 მლ წყალი. აურიეთ მინის წკირით, რომელიც მენზურაში უნდა დარჩეს და მენზურას დააფარეთ საათის მინა. ნაზად მიიყვანეთ ადუღების კონდიციამდე და შეინარჩენეთ დუღილის წერტილი, ვიდრე არ დავინახავთ, რომ ნაცარი აღარ იხსნება (იშლება). გაფილტრეთ ნაცრისგან თავისუფალ ფილტრის ქაღალდზე და შეაგროვეთ ფილტრატი 250 მლ მოცულობის კოლბაში. გარეცხეთ მენზურა და გაფილტრეთ 5 მლ ცხელი 6 მოლი/ლიტრის მარილმჟავით (ამ თავის პუნქტი 3.2) და ორჯერ მდუღარე წყლით. მოცულობითი კოლბა ნიშნულამდე შეავსეთ წყლით (HCl კონცენტრაცია დაახლოებით 0,5 მოლ/ლიტრზე). 5.1.1.2. თუ ფილტრში ნაშთი (ნარჩენები) გამოჩნდება შავად (ნახშირბადი), ისევ ღუმელში მოათავსეთ და კვლავ მოახდინეთ დანაცრება 450-დან 475°C-მდე. ეს დანაცრება, რომელიც მხოლოდ რამდენიმე საათს მოითხოვს (დაახლოებით სამიდან ხუთ საათამდე), სრულდება, როდესაც ნაცარი გახდება თეთრი ან თითქმის თეთრი. ნაშთი (ნარჩენი) იხსნება დაახლოებით 2 მლ მარილმჟავათი (ამ თავის პუნქტი 3.1), ააორთქლეთ გამოშრობამდე და დაუმატეთ 5 მლ 6 მოლი/ლიტრი მარილმჟავა (ამ თავის პუნქტი 3.2). გააცხელეთ, გაფილტრეთ ხსნარი მოცულობის კოლბაში და ნიშნულამდე შევავსეთ წყლით (HCl კონცენტრაცია დაახლოებით 0,5მოლ/ლიტრზე).
შენიშვნები: ა) კვალი ელემენტების განსაზღვრისას მნიშვნელოვანია ფრთხილად ვიყოთ დაბინძურების რისკებთან, განსაკუთრებით თუთის, სპილენძითა და რკინით დაბინძირებისგან. ამის გამო, ნიმუშების მომზადებისას, მოწყობილობა, რომელსაც ვიყენებთ არ უნდა შეიცავდეს ამ ლითონებს. დაბინძურების ზოგადი რისკის შესამცირებლად, იმუშავეთ მტვრისგან თავისუფალ ატმოსფეროში სკრუპულოზურად სუფთა აღჭურვილობითა და ყურადღებით გარეცხილი მინის ჭურჭლით. თუთიის განსაზღვრა განსაკუთრებით მგრძნობიარეა მრავალი სახის დაბინძურების მიმართ, მაგ. მინის ჭურჭლისგან, რეაგენტებისგან, მტვრისგან და ა.შ. ბ) დანაცრებისთვის განკუთვნილი ნიმუშის წონა გამოითვლება ცხოველის საკვების კვალი ელემენტის სავარაუდო შემცველობიდან, გამოსაყენებელი სპექტროფოტომეტრის მგრძნობელობასთან მიმართებაში. კონკრეტული ცხოველის საკვებისთვის, რომელთაც აქვთ ნაკლები კვალი ელემენტი, შეიძლება საჭირო გახდეს 10-დან 20 გ-მდე ნიმუშით დაწყება და საბოლოო ხსნარის მიღება მხოლოდ 100 მლ-მდე. გ) დანაცრება უნდა წარმოებდეს დახურულ ღუმელში, ჰაერის ან ჟანგბადის ინჯექტირების გარეშე. დ) პირომეტრის მიერ მითითებული ტემპერატურა არ უნდა აღემატებოდეს 475oC – ს.
5.1.2. სპექტროფოტომეტრიული განსაზღვრა 5.1.2.1. საკალიბრო ხსნარების მომზადება თითოეული ელემენტის განსაზღვრისათვის, ამ თავის 3.7.1, 3.8.1, 3.9.1 და 3.10.1-ე პუნქტებში მოცემული სტანდარტული სამუშაო ხსნარებიდან მოამზადეთ საკალიბრო ხსნარების სპექტრი (დიაპაზონი) ისე, რომ თითოეული საკალიბრო ხსნარს გააჩნდეს HCl კონცენტრაცია დაახლოებით 0,5 მოლი/ლიტრზე (და (რკინის, მანგანუმისა და თუთიის შემთხვევაში) ლანთანის ქლორიდის კონცენტრაცია, რომელიც ეკვივალენტურია 0,1% La (წონა/მოც).
შერჩეული კვალი ელემენტის კონცენტრაცია უნდა იყოს სპექტროფოტომეტრის გამოყენებული მგრძნობელობის დიაპაზონში. ქვემოთ მოცემულ ცხრილებში მოცემულია, მაგალითად, საკალიბრო ხსნარების ტიპური დიაპაზონის კომპოზიციები; ამასთან, გამოყენებული სპექტროფოტომეტრის ტიპზე და მგრძნობელობაზე დაყრდნობით, შეიძლება საჭირო გახდეს სხვა კონცენტრაციების შერჩევა.
რკინა
სპილენძი
მაგნიუმი
თუთია
5.1.2.2. გამოკვლევისთვის ხსნარის მომზადება
სპილენძის დასადგენად, ამ თავის 5.1.1-ე პუნქტიდან მომზადებული ხსნარი, როგორც წესი შეიძლება გამოყენებულ იქნეს პირდაპირ. საჭიროების შემთხვევაში, რათა მოხდეს მისი კონცეტრაციის საკალიბრო ხსნარების დიაპაზონში მიღება, ალიქვოტის პორცია შეიძლება პიპეტით გადატანილი იქნეს 100 მლ მოცულობის კოლბაში და ნიშნულამდე შეივსოს 0,5 მოლი/ლიტრის მარილმჟავით (ამ თავის პუნქტი 3.3).
რკინის, მანგანუმისა და თუთიის განსაზღვრისთვის, პიპეტით გადაიტანეთ ამ თავის 5.1.1 პუნქტიდან მომზადებული ნიმუშის ალიქვოტის პორცია 100 მლ მოცულობის კოლბაში, დაუმატეთ 10 მლ ლანთანის ქლორიდის ხსნარი (ამ თავის პუნქტი 3.11) და ნიშნულამდე შეავსეთ 0,5 მოლი/ლიტრი მარილმჟავით (ამ თავის პუნქტი 3.3) (იხ. აგრეთვე ამ თავის პუნქტი 8 – დაკვირვება).
5.1.2.3. საკონტროლო ექსპერიმენტი ბრმა (საკონტროლო) ტესტი უნდა მოიცავდეს მომზადების პროცედურაში აღწერილ ყველა ნაბიჯს, გარდა იმ შემთხვევისა, როდესაც მასალის ნიმუში გამოტოვებულია. საკალიბრო ხსნარი „0“ არ უნდა იქნეს გამოყენებული, როგორც საკონტროლო ხსნარი.
5.1.2.4. ატომური აბსორბაციის გაზომვა გაზომეთ საკალიბრო ხსნარების და გამოსაკვლევი ხსნარის ატომური აბსორაბცია ჟანგვითი აცეტილენ-ჟანგბადოვანი ალის გამოყენებით შემდეგ ტალღურ სიგრძეებზე:
თითოეული გაზომვა ოთხჯერ განახორციელეთ.
5.2. ცხოველის მინერალური საკვები
თუ ნიმუში არ შეიცავს ორგანულ ნივთიერებებს, წინასწარი დანაცრება საჭირო არ არის. იმოქმედეთ ისე, როგორც ეს აღწერილია ამ თავის 5.1.1.1. პუნქტში, მეორე აბზაციდან დაწყებული. აორთქლება ფტორწყალბადმჟავასთან შეიძლება იყოს გამოტოვებული.
6. შედეგების დაანგარიშება საკალიბრო მრუდის გამოყენებით, გამოთვალეთ კვალი ელემენტის კონცენტრაცია გამოსაკვლევ ხსნარში და კვალი ელემენტის შედეგი გამოხატეთ მილიგრამებში ნიმუშის თითოეულ კილოგრამზე (ppm).
7. განმეორებადობა ერთი და იმავე ანალიტიკოსის მიერ, ერთსა და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს:
– 5 მგ/კგ-ს აბსოლუტურ მნიშვნელობაში, შესაბამისი კვალი ელემენტის შემცველობით 50 მგ/კგ-მდე; – 10 მგ/კგ-ს უფრო მაღალ მაჩვენებელთან მიმართებაში, როდესაც ამ კვალი ელემენტების შემცველობა 50-დან 100 მგ/კგ-მდეა; – 10 მგ/კგ-ს აბსოლუტურ მნიშვნელობაში, შესაბამისი კვალი ელემენტის შემცველობით 100-დან 200 მგ/კგ-მდე; – 5%-ს უფრო მაღალ მაჩვენებელთან მიმართებაში, როდესაც ამ კვალი ელემენტის შემცველობა 200 მგ/კგ-ზე მეტია.
8. დაკვირვება დიდი რაოდენობით ფოსფატების არსებობამ შეიძლება ხელი შეუშალოს რკინის, მანგანუმის და თუთიის განსაზღვრას. ასეთი ჩარევა უნდა გამოსწორდეს ლანთანის ქლორიდის ხსნარის დამატებით (ამ თავის პუნქტი 3.11). ამასთან, თუ წონის თანაფარდობა ნიმუშში არის Ca+Mg/P>2, გამოსაკვლევ ხსნარში და საკალიბრო ხსნარში ლანთანის ქლორიდის ხსნარის დამატება (ამ თავის პუნქტი 3.11) შეიძლება გამოტოვოთ.
თავი IV. ჰალოფუგინონის განსაზღვრა DL-ტრანს-7-ბრომო-6-ქლორო-3 – [3 – (3-ჰიდროქსი-2-პიპერიდილ) აცეტონილ] -ქინაზოლინ-4 – (3H) -ონის ჰიდრობრომიდი
1. მიზანი და გამოყენების სფერო აღნიშნული მეთოდი საშუალებას იძლევა განვსაზღვროთ ჰალოფუგინონის შემცველობა ცხოველის საკვებში. რაოდენობრივი შეფასების ზღვარი არის 1 მგ/კგ.
2. გამოყენების პრინციპი ცხელი წყლით დამუშავების შემდეგ, ჰალოფუგინონი ექსტრაჰირდება (მიიღება), როგორც თავისუფალი ფუძე ეთილის აცეტატში და შემდეგ ჰიდროქლრიდის სახით ნაწილდება მჟავის წყალხსნარში. ექსტრაქტი იწმინდება იონგაცვლითი ქრომატოგრაფიით. ჰალოფუგინონის შემცველობა განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (HPLC) UV (ულტრაიისფერი) დეტექტორის გამოყენებით.
3. რეაგენტები 3.1. აცეტონიტრილი, ეკვივალენტი HPLC კლასის. 3.2. Amberlite XAD-2. 3.3. ამონიუმის აცეტატი. 3.4. ეთილის აცეტატი. 3.5. ძმარმჟავა, დაკრისტალებული . 3.6. ჰალოფუგინონის სტანდარტული ნივთიერება DL-ტრანს-7-ბრომო-6-ქლორო-3 – [3 – (3-ჰიდროქსი-2-პიპერიდილ) აცეტონილ] -ქინაზოლინ-4 – (3H) -ერთი ჰიდრობრომიდი, E 764) 3.6.1. ჰალოფუგინონის საწყისი სტანდარტული ხსნარი, 1 0 0 მკგ/მლ
0,1 მგ-მდე სიზუსტით, აწონეთ 50 მგ ჰალოფუგინონი (ამ თავის პუნქტი 3.6) 500 მლ გრადირებულ კოლბაში, გახსენით ამონიუმის აცეტატის ბუფერულ ხსნარში (ამ თავის პუნქტი 3.18), ნიშნულამდე შეავსეთ ბუფერული ხსნარით და აურიეთ. ეს ხსნარი მდგრადია სამი კვირის განმავლობაში 5oC ტემპერატურაზე, თუ სიბნელეში ინახება. 3.6.2. საკალიბრო ხსნარები 1,0, 2,0, 3,0, 4,0 და 6,0 მლ საწყისი სტანდარტული ხსნარი გადაიტანეთ 100 მლ გრადურიებული კოლბების სერიაში (ამ თავის პუნქტი 3.6.1). შეავსეთ ნიშნულამდე მობილური ფაზით (ამ თავის პუნქტი 3.21) და აურიეთ. ამ ხსნარებს აქვთ 1,0, 2,0, 3,0, 4,0 და 6,0 მკგ/მლ ჰალოფუგინონის კონცენტრაცია. გამოყენებამდე ხსნარი ახალი მომზადებული უნდა იყოს. 3.7. მარილმჟავა (ρ20 დაახლოებით 1,16 გ/მლ). 3.8. მეთანოლი. 3.9. ვერცხლის ნიტრატი. 3.10. ნატრიუმის ასკორბატი. 3.11. ნატრიუმის კარბონატი. 3.12. ნატრიუმის ქლორიდი. 3.13. EDTA (ეთილენდიამინტეტრაძმარმჟავა, ნატრიუმის მარილი). 3.14. წყალი, HPLC კლასის (ხარისხის) ეკვივალენტი. 3.15. ნატრიუმის კარბონატის ხსნარი, c = 10 გ/100 მლ. 3.16. ნატრიუმის ქლორიდით გაჯერებული ნატრიუმის კარბონატის ხსნარი, c = 5 გ/100 მლ. გახსენით 50 გ ნატრიუმის კარბონატი (ამ თავის პუნქტი 3.11) წყალში, განაზავეთ 1 ლიტრამდე და დაამატეთ ნატრიუმის ქლორიდი (ამ თავის პუნქტი 3.12) სანამ ხსნარი არ გაჯერდება. 3.17. მარილმჟავა, დაახლოებით 0,1 მოლი/ლიტრი. განზავდეს 10 მლ HCI (ამ თავის პუნქტი 3.7) წყლით 1 ლიტრამდე. 3.18. ამონიუმის აცეტატის ბუფერული ხსნარი, დაახლოებით 0,25 მოლი/ლიტრი. გააზავეთ 19,3 გ ამონიუმის აცეტატი (ამ თავის პუნქტი 3.3) და 30 მლ ძმარმჟავა (ამ თავის პუნქტი 3.5) წყალში (ამ თავის პუნქტი 3.14) 1 ლ-მდე. 3.19. Amberlite XAD-2 ფისის მომზადება. გარეცხეთ შესაბამისი რაოდენობით ამბერლიტი (ამ თავის პუნქტი 3.2) წყლით, სანამ ქლორიდის ყველა იონი არ მოცილდება, როგორც ეს მითითებულია ვერცხლის ნიტრატის (ამ თავის პუნქტი 3.20) ტესტით, რომელიც ჩატარებულია გამოყენებული (ნახმარი) წყალის ფაზაზე. შემდეგ ფისი გარეცხეთ 50 მლ მეთანოლით (ამ თავის პუნქტი 3.8), მოაცილეთ მეთანოლი და შეინახეთ ფისი ახალი მეთანოლის ქვეშ. 3.20. ვერცხლის ნიტრატის ხსნარი, დაახლოებით 0,1 მოლი/ლიტრი. გახსენით 0,17 გ ვერცხლის ნიტრატი (ამ თავის პუნქტი 3.9) 10 მლ წყალში. 3.21. HPLC მობილური ფაზა. აურიეთ 500 მლ აცეტონიტრილი (ამ თავის პუნქტი 3.1) 300 მლ ამონიუმის აცეტატის ბუფერულ ხსნართან (ამ თავის პუნქტი 3.18) და 1 200 მლ წყალთან (ამ თავის პუნქტი 3.14). pH დააკორექტირეთ 4,3-ზე ძმარმჟავას გამოყენებით (ამ თავის პუნქტი 3.5). გაფილტრეთ 0,22 მკმ ფილტრით (ამ თავის პუნქტი 4.8) და მოახდინეთ ხსნარის დეგაზირება (მაგ. ულტრასონიფიკაციით 10 წუთის განმავლობაში). ეს ხსნარი სტაბილურია ერთი თვის განმავლობაში, თუ ინახება სიბნელეში, დახურულ ჭურჭელში. 4. აპარატურა 4.1. ულტრაბგერითი აბაზანა 4.2. როტორული ამაორთქლებელი ვიდეო ჩამწერით 4.3. ცენტრიფუგა 4.4. HPLC მოწყობილობა ცვლადი ტალღის ულტრაიისფერი დეტექტორით ან დიოდურ-მატრიცული დეტექტორით 4.4.1. თხევადი ქრომატოგრაფიული სვეტი, 300 მმ x 4 მმ, C 18, 10 მკმ შეფუთვა, ან ეკვივალენტური სვეტი 4.5. მინის სვეტი (300 მმ x 10 მმ) დამონტაჟებული სინთეზური მინის ფილტრით და საცობით 4.6. მინის ბოჭკოვანი ფილტრები, დიამეტრი 150 მმ 4.7. მემბრანული ფილტრები, 0,45 მკმ 4.8. გარსის ფილტრები, 0,22 მკმ
5. გამოყენების პროცედურა შენიშვნა: ჰალოფუგინონი, როგორც თავისუფალი ფუძე, არასტაბილურია ტუტე და ეთილის აცეტატის ხსნარებში. იგი არ უნდა დარჩეს ეთილის აცეტატში 30 წუთზე მეტი ხნის განმავლობაში.
5.1. ზოგადი პრინციპი 5.1.1. უნდა ჩატარდეს ცხოველის საკონტროლო საკვების გამოკვლევა, რათა გამოირიცხოს ჰალოფუგინონისა და ხელშემშლელი ნივთიერებების არსებობა.
5.1.2. აღდგენითი ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების გამოკვლევის გზით, რომელიც გაჯერებულია (გამდიდრებულია) ჰალოფუგინონის რაოდენობის დამატებით, მსგავსად იმისა, როგორც ეს მოცემულია ნიმუშში. 3 მგ/კგ დონეზე გაჯერებისთვის (კონცენტრაციის მისაღებად), 10 გ ცხოველის საკონტროლო საკვებს დაუმატეთ 300 მკლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.6.1), აურიეთ და გააჩერეთ 10 წუთი, სანამ გადახვალთ ექსტრაჰირების ეტაპზე (ამ თავის პუნქტი 5.2).
შენიშვნა: ამ მეთოდის მიზნებისათვის ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის ტიპის მსგავსი და ანალიზისას, ჰალოფუგინონი არ უნდა ვლინდებოდეს.
5.2 ექსტრაჰირება აწონეთ 10 გ მომზადებული ნიმუში, 0,1 გ სიზუსტით, 200 მლ ცენტრიფუგის მილში, დაამატეთ 0,5 გ ნატრიუმის ასკორბატი (ამ თავის პუნქტი 3.10), 0,5 გ EDTA (ამ თავის პუნქტი 3.13) და 20 მლ წყალი, და აურიეთ. მოათავსეთ მილი (ტუბი) 5 წუთის განმავლობაში წყლის აბაზანაში (80oC). ოთახის ტემპერატურაზე გაგრილების შემდეგ, დაამატეთ 20 მლ ნატრიუმის კარბონატის ხსნარი (ამ თავის პუნქტი 3.15) და აურიეთ. დაუყოვნებლივ დაამატეთ 100 მლ ეთილის აცეტატი (ამ თავის პუნქტი 3.4) და ხელით ენერგიულად შეანჯღრიეთ 15 წამის განმავლობაში. შემდეგ მოათავსეთ მილი სამი წუთის განმავლობაში ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.1) და მოუშვით საცობი. მოახდინეთ ცენტრიფუგირება ორი წუთის განმავლობაში და ეთილის აცეტატის ფაზის დეკანტირება მინის ბოჭკოვანი ფილტრის საშუალებით (ამ თავის პუნქტი 4.6), 500-მლ-იანი გამყოფი ძაბრით. გაიმეორეთ ნიმუშის ექსტრაჰირება 100 მლ ეთილის აცეტატის მეორე პორციით. კომბინირებული ექსტრაქტები ერთი წუთის განმავლობაში გარეცხეთ 50 მლ ნატრიუმის ქლორიდის ნატრიუმის კარბონატის გაჯერებული ხსნარით (ამ თავის პუნქტი 3.16) და მოაცილეთ წყლის ფენა.
მოახდინეთ ორგანული ფენის 1 წთ-ში ექსტრაჰირება 50 მლ მარილმჟავასთან (ამ თავის პუნქტი 3.17). მჟავას ქვედა ფენა გაატარეთ 250-მლ-იან გამყოფ ძაბრში. მოახდინეთ ორგანული ფენის 1,5 წუთის განმავლობაში ხელახალი ექსტრაჰირება შემდგომი 50 მლ ჰიდროქლორიდის მჟავით და შეურიეთ პირველ ექსტრაქტს. გარეცხეთ კომბინირებული მჟავას ექსტრაქტები დაახლოებით 10 წამის წრიული მოძრაობებით 10 მლ ეთილის აცეტატით (ამ თავის პუნქტი 3.4). რაოდენობრივად გადაიტანეთ წყლის ფენა 250 მლ მრგვალძირიან კოლბაში და მოაცილეთ ორგანული ფაზა. ააორთქლეთ დარჩენილი ეთილის აცეტატის მჟავა ხსნარიდან ვიდეო ჩამწერის მქონე როტორული ამაორთქლებელის გამოყენებით (ამ თავის პუნქტი 4.2). წყლის აბაზანის ტემპერატურა არ უნდა აღემატებოდეს 40oC-ს. დაახლოებით 25 მბ-ის ვაკუუმის პირობებში, 5 წუთის განმავლობაში ამოიღება დანარჩენი ეთილის აცეტატი, 38oC ტემპერატურაზე.
5.3. გაწმენდა 5.3.1. ამბერლიტის სვეტის მომზადება თითოეული ნიმუშის ექსტრაქტისთვის მზადდება XAD-2 სვეტი. გადაიტანეთ 10 გ მომზადებული ამბერლიტი (ამ თავის პუნქტი 3.19) მინის სვეტში (ამ თავის პუნქტი 4.5) მეთანოლთან (ამ თავის პუნქტი 3.8). ფისის ზედა შრეზე (ბალიშზე) მოათავსეთ მინაბოჭკოვანი ბამბის საცობი. სვეტიდან გადაწურეთ მეთანოლი და ჩამორეცხეთ ფისი 100 მლ წყლით, შეაჩერეთ ნაკადი, როცა სითხე მიაღწევს ფისის ზედა ფენას (ბალიშს).
გამოიყენებამდე, დაიცადეთ 10 წუთი, რათა სვეტი გათანაბრდეს. არასოდეს დაუშვათ სვეტის გამოშრობა.
5.3.2. ნიმუშის გაწმენდა ექსტრაქტი (ამ თავის პუნქტი 5.2) რაოდენობრივად გადაიტანეთ მომზადებული ამბერლიტის სვეტის (ამ თავის პუნქტი 5.3.1) ზედა ნაწილში და გამოდევნეთ ელუატი. ელუციის სიჩქარე არ უნდა აღემატებოდეს 20 მლ/წთ. ჩამორეცხეთ მრგვალძირიანი კოლბა 20 მლ მარილმჟავით (ამ თავის პუნქტი 3.17) და გამოიყენეთ იგი ფისოვანი სვეტის გასარეცხად. დარჩენილი მჟავას ხსნარი გაანიავეთ ჰაერის ნაკადით. მოაცილეთ სარეცხი სითხე. სვეტს დაუმატეთ 100 მლ მეთანოლი (ამ თავის პუნქტი 3.8) და დაელოდეთ 5-დან 10 მლ გასუფთავებას, შეაგროვეთ ელუატი 250 მლ მრგვალძირიან კოლბაში. დარჩენილი მეთანოლი დატოვეთ 10 წუთის განმავლობაში, რათა გათანაბრდეს ფისთან და გააგრძელეთ ელუცია სიჩქარეზე, რომელიც არ აღემატება 20 მლ/წთ-ს, თან აწარმოეთ ელუატის შეგროვება იმავე მრგვალძირიან კოლბაში. მეთანოლი აორთქლეთ ვიდეო ჩამწერის მქონე როტორულ ამაორთქლებელზე (ამ თავის პუნქტი 4.2), წყლის აბაზანის ტემპერატურა არ უნდა აღემატებოდეს 40oC-ს. ნაშთი (ნარჩენები) რაოდენობრივად გადაიტანეთ 10 მლ დაკალიბრებულ კოლბაში მობილური ფაზის გამოყენებით (ამ თავის პუნქტი 3.21). შეავსეთ ნიშნულამდე მობილური ფაზით და აურიეთ. ალიქვოტი იფილტრება მემბრანული ფილტრის საშუალებით (ამ თავის პუნქტი 4.7). შეინახეთ ეს ხსნარი HPLC განსაზღვრისთვის (ამ თავის პუნქტი 5.4).
5.4. HPLC განსაზღვრა
5.4.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები, სხვა პირობები შეიძლება გამოყენებულ იქნეს, თუ ისინი იძლევიან ეკვივალენტურ შედეგებს:
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, რამდენჯერმე მოახდინეთ 3,0 მკგ/მლ შემცველობის მქონე საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.6.2), სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები (ან ფართობები) და შეკავების სტაბილური დრო.
5.4.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.6.2) და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი, საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბსცისასი.
5.4.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის ინჯექტირება (ამ თავის პუნქტი 5.3.2), იმავე მოცულობის გამოყენებით, რაც აღებული იქნა საკალიბრო ხსნარებისთვის და განსაზღვრეთ ჰალოფუგინონის მწვერვალების საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების გაანგარიშება განსაზღვრეთ ნიმუშის ხსნარის კონცენტრაცია მკგ/მლ-ში, ნიმუშის ხსნარის ჰალოფუგინონის მწვერვალების საშუალო სიმაღლიდან (ფართობიდან) საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.4.2).
ჰალოფუგინონის ნიმუშის შემცველობა w (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის ჰალოფუგინონის კონცენტრაცია მკგ/მლ-ში; m = გამოსაკვლევი პორციის წონა გრამებში.
7. შედეგების ვალიდაცია 7.1. იდენტიფიკაცია გამოსაკვლევი ნივთიერების (ანალიტის) იდენტურობა შეიძლება დადასტურდეს თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც ხდება ნიმუშის ექსტრაქტის სპექტრებისა და 6,0 მკგ/მლ-ის შემცველობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.6.2) შედარება.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი გამდიდრებულია შესაბამისი რაოდენობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.6.2) დამატებით. ჰალოფუგინონის დამატებული რაოდენობა უნდა იყოს ჰალოფუგინონის სავარაუდო რაოდენობის მსგავსი, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ ჰალოფუგინონის პიკის სიმაღლე. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს თავდაპირველი სიგანის ± 10%-ის ფარგლებში.
7.1.2. გამოვლენა დიოდური მატრიცით
შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით: ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბსორბაციის ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, ინდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცული გამოვლენისთვის ეს ჩვეულებრივ ±2 ნმ-ის ფარგლებშია; ბ) 225-დან 300 ნმ-მდე შუალედში ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია ქრომატოგრამიის პიკის სიმაღლეზე, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100%-მდე შედარებითი შთანთქმის (აბსორბაციის) დიაპაზონში. ეს კრიტერიუმი შესაბამისობაშია მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და, როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული გამოსაკვლევი ნივთიერების (ანალიტის) შთანთქმის 15% -ს; გ) 225-დან 300 ნმ-მდე შუალედში ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის (აბსორბციის) 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15%-ს. თუ რომელიმე ამ კრიტერიუმებიდან არ სრულდება, გამოსაკვლევი ნივთიერების (ანალიტის) არსებობა არ დადასტურდება.
7.2. განმეორებადობა ერთსა და იმავე ნიმუშზე ჩატარებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 0,5 მგ/კგ ჰალოფუგინონის შემცველობის 3 მგ/კგ-მდე.
7.3. აღდგენა გამდიდრებული საკონტროლო ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 80%.
8. ერთობლივი კვლევის შედეგები
რვა ლაბორატორიის მიერ ჩატარებული ერთობლივი კვლევისას გამოკვლეულ იქნა სამი ნიმუში.
შედეგები
ND = არ არის გამოვლენილი; S R = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; CV R = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი (%); Rec. = აღდგენა (%).
თავი V რობენიდინის განსაზღვრა 1,3-ბის [(4-ქლორობენზილიდინი) ამინო] გუანიდინი – ჰიდროქლორიდი
1. მიზანი და გამოყენების სფერო ეს მეთოდი შესაძლებელს ხდის რობენიდინის შემცველობის განსაზღვრას ცხოველის საკვებში. რაოდენობრივი შეფასების ზღვარი არის 5 მგ/კგ.
2. გამოყენების პრინციპი ნიმუში ექსტრაჰირებულია მჟავაშეზავებული მეთანოლით. ექსტრაქტს აშრობენ და ალიქვოტის პორციას ასუფთავებენ ალუმინის ოქსიდის სვეტზე. რობენიდინი ირეცხება სვეტიდან მეთანოლით, კონცენტრირდება და ისხმება, მობილური ფაზით, შესაბამის მოცულობამდე. რობენიდინის შემცველობა განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (HPLC), UV-ის (ულტრაიისფერის) დეტექტორის საშუალებით.
3. რეაგენტები 3.1. მეთანოლი. 3.2. მჟავაშეზავებული მეთანოლი. გადაიტანეთ 4,0 მლ მარილმჟავა (ρ20 = 1,18 გ/მლ) 500 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე მეთანოლით (ამ თავის პუნქტი 3.1) და აურიეთ. გამოყენებამდე ეს ხსნარი უნდა იყოს ახალი მომზადებული. 3.3. აცეტონიტრილი, HPLC კლასის ეკვივალენტური. 3.4. მოლეკულური საცერი. ტიპი 3A, 8 დან 12 ქსელის კვანძები (1,6-2,5 მმ კვანძები, კრისტალური ალუმინო-სილიკატი, ფორების დიამეტრი 0,3 მმ). 3.5. ალუმინის ოქსიდის მჟავას აქტივობის დონე I, სვეტის ქრომატოგრაფიისთვის. 100 გ ალუმინის ოქსიდი გადაიტანეთ შესაბამის ჭურჭელში და დაამატეთ 2,0 მლ წყალი. გაუკეთეთ საცობი და შეანჯღრიეთ დაახლოებით 20 წუთი. შეინახეთ კარგად დახურულ ჭურჭელში. 3.6. კალიუმის დიჰიდროფოსფატის ხსნარი, c = 0,025 მოლი/ლიტრი. გახსენით 3,40 გ კალიუმის დიჰიდროფოსფატი წყალში (HPLC კლასის) 1 000 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე და აურიეთ. 3.7. დი-ნატრიუმის წყალბადის ფოსფატის ხსნარი, c = 0,025 მოლი/ლიტრი. 1-ლიტრიან გრადირებულ კოლბაში გახსენით 3,55 გ უწყლო (ან 4,45 გ დიჰიდრატი ან 8,95 გ დოდეკაჰიდრატი) დი-ნატრიუმის წყალბადის ფოსფატი წყალში (HPLC კლასის ეკვივალენტი), შეავსეთ ნიშნულამდე და აურიეთ. 3.8. HPLC მობილური ფაზა. შეურიეთ შემდეგი რეაგენტები:
გაფილტრეთ 0,22 მკმ ფილტრით (ამ თავის პუნქტი 4.6) და მოახდინეთ ხსნარის დეგაზირება (მაგ. ულტრასონიფიკაციით 10 წუთის განმავლობაში). 3.9. სტანდარტული ნივთიერება. სუფთა რობენიდინი: 1,3-bis [(4-ქლორბენზილიდინი) ამინო] გუანიდინი – ჰიდროქლორიდი. 3.9.1. რობენიდინის საწყისი სტანდარტული ხსნარი: 300 მკგ/მლ აწონეთ 0,1 მგ სიზუსტით, 30 მგ რობენიდინის სტანდარტული ნივთიერება (ამ თავის პუნქტი 3.9). გახსენით მჟავაშერეულ მეთანოლში (ამ თავის პუნქტი 3.2) 100 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე იმავე გამხსნელით და აურიეთ. კოლბა შეახვიეთ ალუმინის ფოლგით და შეინახეთ ბნელ ადგილას. 3.9.2. რობენიდინის შუალედური სტანდარტული ხსნარი: 12 მკგ/მლ 10,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.9.1) გადაიტანეთ 250-მლ-იან გრადირებულ კოლბაში, შეავსეთ მობილური ფაზით (ამ თავის პუნქტი 3.8) და აურიეთ. კოლბა შეახვიეთ ალუმინის ფოლგით და შეინახეთ ბნელ ადგილას.
3.9.3. საკალიბრო ხსნარები 50 მლ დაკალიბრებული კოლბების სერიაში გადაიტანეთ 5,0, 10,0, 15,0, 20,0 და 25,0 მლ შუალედური სტანდარტული სნარი (ამ თავის პუნქტი 3.9.2). შეავსეთ ნიშნულამდე მობილური ფაზით (ამ თავის პუნქტი 3.8) და აურიეთ. ეს ხსნარები შეესაბამება 1,2, 2,4, 3,6, 4,8 და 6,0 მკგ/მლ რობენიდინს. გამოყენებამდე ეს ხსნარები უნდა იყოს ახალი მომზადებული. 3.10. HPLC კლასის ეკვივალენტური წყალი.
4. აპარატურა 4.1. მინის სვეტი. დამზადებულია ქარვის მინისგან, რომელსაც გააჩნია საცობი და რეზერვუარი დაახლოებით 150 მლ მოცულობის, შიდა დიამეტრით 10-დან 15 მმ-მდე, სიგრძით 250 მმ. 4.2. მექანიკური სადღვები ან მაგნიტური სათქვეფელა. 4.3. როტორული ამაორთქლებელი ვიდეოჩამწერით. 4.4. HPLC მოწყობილობა ცვლადი ტალღის სიგრძის ულტრაიისფერი დეტექტორით ან დიოდური მატრიცის დეტექტორით, რომელიც მუშაობს 250-დან 400 ნმ დიაპაზონში. 4.4.1. თხევადი ქრომატოგრაფიული სვეტი: 300 მმ x 4 მმ, C 18 10 მკმ შეფუთვა ან ეკვივალენტი. 4.5. მინის ბოჭკოვანი ფილტრის ქაღალდი (Whatman GF/A ან ეკვივალენტი). 4.6. მემბრანული ფილტრები, 0,22 მკმ. 4.7. მემბრანული ფილტრები, 0,45 მკმ.
5. გამოყენების პროცედურა
შენიშვნა: რობენიდინი მგრძნობიარეა შუქზე. ქარვის მინის ჭურჭელი გამოყენებული უნდა იქნეს ყველა ოპერაციაში.
5.1. ზოგადი პრონციპები
5.1.1. კვლევა უნდა ჩატარდეს ცხოველის საკონტროლო საკვების შესამოწმებლად, რათა გამოირიცხოს რობენიდინისა და დამაბრკოლებელი ნივთიერებების არსებობა.
5.1.2. აღდგენის ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების (ამ თავის პუნქტი 5.1.1) გამოკვლევით, რომელიც გამდიდრებულია რობენიდინის რაოდენობის დამატებით, რაც ნიმუშში არსებულის იდენტურია. 60 მგ/კგ დონეზე გამდიდრების მიზნით, 3,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.9.1) გადაიტანეთ 250 მლ კონუსურ კოლბაში. აორთქლეთ გამხსნელი დაახლოებით 0,5 მლ-მდე აზოტის ნაკადში. დაუმატეთ 15 გ ცხოველის საკონტროლო საკვები, აურიეთ და დაელოდეთ 10 წუთს, სანამ გადახვალთ ექსტრაჰირების ეტაპზე (ამ თავის პუნქტი 5.2).
შენიშვნა: ამ მეთოდის მიზნებისათვის ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის მსგავსი და რობენიდინის კვლევისას არ უნდა ვლინდებოდეს.
5.2. ექსტრაჰირება აწონეთ 0,01 გ-მდე სიზუსტით, დაახლოებით 15 გ მომზადებული ნიმუში. გადაიტანეთ 250 მლ კონუსურ კოლბაში და დაუმატეთ 100,0 მლ მჟავაშეზავებული მეთანოლი (ამ თავის პუნქტი 3.2), გაუკეთეთ საცობი და შეანჯღრიეთ ერთი საათით სადღვებზე (ამ თავის პუნქტი 4.2). ხსნარი გაფილტრეთ მინის ბოჭკოვანი ფილტრის ქაღალდის საშუალებით (ამ თავის პუნქტი 4.5) და შეაგროვეთ მთელი ფილტრაცია 150 მლ კონუსურ კოლბაში. დაუმატეთ 7,5 გ მოლეკულური საცერი (ამ თავის პუნქტი 3.4), გაუკეთეთ საცობი და გააჩერეთ ხუთი წუთის განმავლობაში. დაუყოვნებლივ გაფილტრეთ მინის ბოჭკოვანი ფილტრის ქაღალდის საშუალებით. ხსნარი შეინახეთ გაწმენდის ეტაპისთვის (ამ თავის პუნქტი 5.3).
5.3. გაწმენდის პროცედურა
5.3.1. ალუმინის – ოქსიდის სვეტის მომზადება მოათავსეთ მინაბოჭკოვანი ბამბისგან დამზადებული პატარა ზომის საცობი მინის სვეტის ქვედა ბოლოში (ამ თავის პუნქტი 4.1) და მინის ჯოხის გამოყენებით მოახდინეთ მისი ჩატენა. აწონეთ 11,0 გ ალუმინის მზა ოქსიდი (ამ თავის პუნქტი 3.5) და გადაიტანეთ სვეტზე. ზომები უნდა იქნეს მიღებული, რომ ამ ეტაპზე ჰაერის წნევა დაყვანილ იქნეს მინიმუმამდე. ნაზად მიუკაკუნეთ სვეტის დატვირთულ ქვედა ბოლოს, რათა ალუმინის ოქსიდი დაილექოს.
5.3.2. ნიმუშის გაწმენდა პიპეტით სვეტზე გადაიტანეთ 5,0 მლ ნიმუშის ექსტრაქტი, რომელიც მომზადებულია ამ თავის 5.2-ე პუნქტში მითითებული წესის მიხედვით. პიპეტის წვერი მოათავსეთ სვეტის კედელთან და მიეცით ხსნარს საშუალება, რომ მოხდეს მისი შეწოვება ალუმინის ოქსიდის მიერ. რობენიდინი გამოდევნეთ სვეტიდან 100 მლ მეთანოლის გამოყენებით (ამ თავის პუნქტი 3.1), 2-დან 3 მლ/წთ სიჩქარით და შეაგროვეთ ელუატი 250 მლ მრგვალძირიან კოლბაში. აორთქლეთ მეთანოლის ხსნარი მშრალ მდგომარეობაში 40°C ტემპერატურაზე შემცირებული წნევის ქვეშ ვიდეოჩამწერი როტორული ამაორთქლებელის საშუალებით (მ თავის პუნქტი 4.3). ნაშთი (ნარჩენი) ხელახლა გახსენით 3-დან 4 მლ-მდე მობილურ ფაზაში (ამ თავის პუნქტი 3.8) და რაოდენობრივად გადაიტანეთ 10 მლ გრადირებულ კოლბაში. ჩამორეცხეთ კოლბა 1-2 მლ მობილური ფაზის რამდენიმე პორციით და გადაიტანეთ ეს ჩამონარეცხი გრადირებულ კოლბაში. შეავსეთ ნიშნულამდე იმავე გამხსნელით და აურიეთ. ალიქვოტის პორცია გაფილტრეთ 0,45 მკმ მემბრანულ ფილტრში (ამ თავის პუნქტი 4.7). ხსნარი შეინახეთ HPLC განსაზღვრისთვის (ამ თავის პუნქტი 5.4).
5.4. HPLC განსაზღვრა
5.4.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან: – თხევადი ქრომატოგრაფიული სვეტი (ამ თავის პუნქტი 4.4.1) – HPLC მობილური ფაზა (ამ თავის პუნქტი 3.8) – ნაკადის სიჩქარე: 1,5-დან 2 მლ/წთ. – გამოვლენის ტალღის სიგრძე: 317 ნმ – ინჯექტირების მოცულობა: 20-დან 50 მკლ-მდე.
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, რამდენჯერმე მოახდინეთ 3,6 მკგ/მლ შემცველობის მქონე საკალიბრო ხსნარის (ამ თავის პუნქტი 3.9.3) ინჯექტირება, სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები და შეკავების სტაბილური დრო.
5.4.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის (ამ თავის 3.9.3) ინჯექტირება და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი, საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბცისასი.
5.4.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის (ამ თავის პუნქტი 5.3.2) ინჯექტირება, იმავე მოცულობის გამოყენებით, რაც აღებული იქნა საკალიბრო ხსნარებისთვის და განსაზღვრეთ რობენიდინის პიკის საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების გაანგარიშება განსაზღვრეთ ნიმუშის ხსნარის კონცენტრაცია მკგ/მლ-ში, ნიმუშის ხსნარის რობენიდინის პიკის საშუალო პიკის სიმაღლიდან (ფართობიდან) საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.4.2). რობენიდინის ნიმუშის შემცველობა w (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის რობენიდინის კონცენტრაციას მკგ/მლ-ში; m = გამოსაკვლევი პორციის წონა გრამებში.
7. შედეგების ვალიდაცია
7.1. იდენტიფიკაცია გამოსაკვლევი ნივთიერების (ანალიტის) იდენტურობა შესაძლება დადასტურდეს თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც ხდება ნიმუშის ექსტრაქტის სპექტრებისა და 6 მკგ/მლ-ის შემცველობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.9.3) შედარება.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი გამდიდრებულია შესაბამისი რაოდენობის საკალიბრო ხსნარის დამატებით (ამ თავის პუნქტი 3.9.3). რობენიდინის დამატებული რაოდენობა უნდა იყოს რობენიდინის სავარაუდო რაოდენობის მსგავსი, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ რობენიდინის პიკის სიმაღლე. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს თავდაპირველი სიგანის 10%-ში ფარგლებში.
7.1.2. დიოდური მატრიცით გამოვლენა შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით: ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბსორბაციის ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, იდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცის გამოვლენისთვის ეს ჩვეულებრივ 2 ნმ-ის ფარგლებშია; ბ) 250-დან 400 ნმ-მდე შუალედში ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100%-მდე ფარდობითი აბსორბაციის დიაპაზონში. ეს კრიტერიუმი შესაბამისობაშია მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული გამოსაკვლევი ნივთიერების (ანალიტის) შთანთქმის 15%-ს; გ) 250-დან 400 ნმ-მდე შუალედში, ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და, როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15%-ს.
თუ რომელიმე ამ კრიტერიუმებიდან არ სრულდება, გამოსაკვლევი ნივთიერების (ანალიტის) არსებობა არ დადასტურდება.
7.2. განმეორებადობა ერთი და იმავე ნიმუშზე ჩატარებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 15 მგ/კგ-ზე მეტი რობენიდინის შემცველობის უფრო მაღალი შედეგის 10%-ს.
7.3. აღდგენა გამდიდრებული საკონტროლო ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 85%.
8. ერთობლივი კვლევის შედეგები ჩატარდა EC-ის ერთობლივი კვლევა, რომელშიც 12-მა ლაბორატორიამ გამოიკვლია შინაური ფრინველისა და კურდღლის ცხოველის საკვების ოთხი ნიმუში, ფქვილში ან გრანულირებული სახით. თითოეულ ნიმუშზე ჩატარდა განმეორებითი (დუბლიკატი) კვლევა. შედეგები მოცემულია ცხრილში:
შედეგები
sr = განმეორებადობის სტანდარტული გადახრა; CVr = განმეორებადობის ვარიაციის კოეფიციენტი, %; SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი %.
თავი VI დიკლაზურილის განსაზღვრა (+)-4-ქლორფენილ [2,6-დიქლორო-4-(2,3,4,5-ტეტრაჰიდრო-3,5-დიოქსო-1,2,4-ტრიაზინ-2-ილ) ფენილ] აცეტონიტრილი
1. მიზანი და გამოყენების სფერო აღნიშნული მეთოდი საშუალებას იძლევა განვსაზღვროთ დიკლაზურილის შემცველობა ცხოველის საკვებსა და პრემიქსებში. გამოვლენის ზღვარი არის 0,1 მგ/კგ, რაოდენობრივი შეფასების ზღვარი არის 0,5 მგ/კგ.
2. გამოყენების პრინციპი შიდა სტანდარტის დამატების შემდეგ, ნიმუში მიიღება მჟავაშეზავებული მეთანოლით. ცხოველის საკვების მისაღებად, ალიქვოტის ექსტრაქტის პორცია იწმინდება C18 მყარი ფაზის ექსტრაქციის კარტრიჯით. დიკლაზურილი კატრიჯიდან იწმინდება მჟავაშეზავებული მეთანოლისა და წყლის ნარევით. აორთქლების შემდეგ ნაშთი (ნარჩენები) იხსნება DMF/წყალში. პრემიქსებისთვის, ექსტრაქტი ორთქლდება და ნაშთი (ნარჩენები) იხსნება DMF/წყალში. დიკლაზურილის შემცველობა განისაზღვრება ტერნარული გრადიენტის შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (HPLC) UV დეტექტორის გამოყენებით.
3. რეაგენტები 3.1. წყალი, HPLC ხარისხის ეკვივალენტური 3.2. ამონიუმის აცეტატი 3.3. ტეტრაბუტილამონიუმის წყალბადის სულფატი (TBHS) 3.4. აცეტონიტრილი, HPLC ხარისხის ეკვივალენტური 3.5. მეთანოლი, HPLC ხარისხის ეკვივალენტური 3.6. N, N – დიმეთილფორმამიდი (DMF) 3.7. მარილმჟავა, ρ20 = 1,19 გ/მლ 3.8. სტანდარტული ნივთიერება: დიკლაზურილ II-24: (+)-4-ქლორფენილ [2,6-დიქლორო-4-(2,3,4,5-ტეტრაჰიდრო-3,5-დიოქსო-1,2,4-ტრიაზინ-2)-ილ)ფენილ] აცეტონიტრილი გარანტირებული სისუფთავით, E771 3.8.1. დიკლაზურილის საწყისი სტანდარტული ხსნარი, 500 მკგ/მლ აწონეთ 0,1 მგ სიზუსტით, 25 მგ დიკლაზურილის სტანდარტული ნივთიერება (ამ თავის პუნქტი 3.8) 50 მლ გრადირებულ კოლბაში. გახსენით DMF (ამ თავის პუნქტი 3.6), შეავსეთ ნიშნულამდე DMF (ამ თავის პუნქტი 3.6) და აურიეთ. შემოახვიეთ კოლბას ალუმინის ფოლგა ან გამოიყენეთ ქარვის კოლბა და შეინახეთ მაცივარში. ≤4oC ტემპერატურაზე ხსნარი სტაბილურია 1 თვის განმავლობაში.
3.8.2. დიკლაზურილის სტანდარტული ხსნარი, 50 მკგ/მლ გადაიტანეთ 5,00 მლ საწყისი სტანდარტული ხსნარი (3.8.1) 50 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე DMF (3.6) და აურიეთ. შემოახვიეთ კოლბას ალუმინის ფოლგა ან გამოიყენეთ ქარვის კოლბა და შეინახეთ მაცივარში. ≤ 4oC ტემპერატურაზე ხსნარი სტაბილურია 1 თვის განმავლობაში. 3.9. შიდა სტანდარტული ნივთიერება: 2,6 დიქლორო-α – (4-ქლოროფენილ)-4-(4,5 დიჰიდრო-3,5-დიოქსო-1,2,4-ტრიაზინ-2 (3H)-ილ) α-მეთილბენზოლი-აცეტონიტრილი. 3.9.1. შიდა საწყისი სტანდარტული ხსნარი, 500 მკგ/მლ აწონეთ 0,1 მგ სიზუსტით, 25 მგ შიდა სტანდარტის ნივთიერება (ამ თავის პუნქტი 3.9) 50 მლ გრადირებულ კოლბაში. გახსენით DMF-ში (ამ თავის პუნქტი 3.6), შეავსეთ ნიშნულამდე DMF-ით (ამ თავის პუნქტი 3.6) და აურიეთ. შემოახვიეთ კოლბა ალუმინის ფოლგით ან გამოიყენეთ ქარვის კოლბა და შეინახეთ მაცივარში. ≤4oC ტემპერატურაზე ხსნარი სტაბილურია 1 თვის განმავლობაში.
3.9.2. შიდა სტანდარტული ხსნარი, 50 მკგ/მლ გადაიტანეთ 5,00 მლ შიდა საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.9.1) 50 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე DMF-ით (ამ თავის პუნქტი 3.6) და აურიეთ. შემოახვიეთ კოლბას ალუმინის ფოლგა ან გამოიყენეთ ქარვის კოლბა და შეინახეთ მაცივარში. ≤4oC ტემპერატურაზე ხსნარი სტაბილურია 1 თვის განმავლობაში.
3.9.3. შიდა სტანდარტული ხსნარი პრემიქსებისთვის, p/1000 მგ/მლ (p = დიკლაზურილის ნომინალური შემცველობა პრემიქსებში მგ/კგ-ში) აწონეთ შიდა სტანდარტული ხსნარი 0,1 მგ/10 მგ სიზუსტით, 100 მლ გრადირებულ კოლბაში, გახსენით DMF-ში (ამ თავის პუნქტი 3.6) ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.6), შეავსეთ ნიშნულამდე DMF-ით და აურიეთ. შემოახვიეთ კოლბა ალუმინის ფოლგა ან გამოიყენეთ ქარვის კოლბა და შეინახეთ მაცივარში. ≤ 4oC ტემპერატურაზე ხსნარი სტაბილურია 1 თვის განმავლობაში. 3.10. საკალიბრო ხსნარი, 2 მკგ/მლ. პიპეტით შეყავთ 2,00 მლ დიკლაზურილის სტანდარტული ხსნარი (ამ თავის პუნქტი 3.8.2) და 2,00 მლ შიდა სტანდარტული ხსნარი (ამ თავის პუნქტი 3.9.2) 50 მლ გრადირებულ კოლბაში. დაამატეთ 16 მლ DMF (ამ თავის პუნქტი 3.6), შეავსეთ ნიშნულამდე წყლით და აურიეთ. ეს ხსნარი, გამოყენებამდე უნდა იყოს ახალი მომზადებული. 3.11. C18 მყარი ფაზის ექსტრაქციის კარტრიჯი, მაგ. Bond Elut, ზომა: 1 კუბ., სანტ., სორბენტის წონა: 100 მგ. 3.12. ექსტრაჰირების გამხსნელი: მჟავაშეზავებული მეთანოლი. პიპეტით გადაიტანეთ 5,0 მლ მარილმჟავა (ამ თავის პუნქტი 3.7) 1 000 მლ მეთანოლში (ამ თავის პუნქტი 3.5) და აურიეთ. 3.13. მობილური ფაზა HPLC-სთვის 3.13.1. ელუენტი A: ამონიუმის აცეტატი – ტეტრაბუტილამონიუმის წყალბადის სულფატის ხსნარი. 5 გ ამონიუმის აცეტატი (ამ თავის პუნქტი 3.2) და 3,4 გ TBHS (ამ თავის პუნქტი 3.3) გახსენით 1 000 მლ წყალში (ამ თავის პუნქტი 3.1) და აურიეთ. 3.13.2. ელუენტი B: აცეტონიტრილი (ამ თავის პუნქტი 3.4). 3.13.3. ელუენტი C: მეთანოლი (ამ თავის პუნქტი 3.5).
4. აპარატურა 4.1. მექანიკური სადღვები 4.2. სამსაფეხურიანი HPLC მოწყობილობა 4.2.1. თხევადი ქრომატოგრაფიული სვეტი, Hypersil ODS, 3 მკმ შეფუთვა, 100 მმ x 4.6 მმ ან ეკვივალენტი 4.2.2. UV-ს დეტექტორი ტალღის ცვალებადი რეგულირების ან დიოდური მატრიცის დეტექტორით 4.3. როტორული ამაორთქლებელი ვიდეოჩამწერი აპარატურით 4.4. მემბრანული ფილტრი, 0.45 მკმ 4.5. ვაკუუმის გამანაწილებელი 4.6. ულტრაბგერითი აბაზანა
5. გამოყენების პროცედურა
5.1. ზოგადი პრინციპები 5.1.1. ცხოველის საკონტროლო საკვები კვლევა უნდა ჩატარდეს ცხოველის საკონტროლო საკვების შესამოწმებლად, რათა გამოირიცხოს დიკლაზურილისა და ხელშემშლელი ნივთიერებების არსებობა. ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის ტიპის მსგავსი და კვლევის დროს არ უნდა გამოვლინდეს დიკლაზურილი ან ხელშემშლელი ნივთიერებები.
5.1.2. აღდგენის ტესტი აღდგენის ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების კვლევით, რომელიც გამდიდრებულ იქნა დიკლაზურილის იმ რაოდენობის დამატებით, როგორც ეს წარმოდგენილია ნიმუშში. 1 მგ/კგ დონეზე გამდიდრებისთვის დაამატეთ 0,1 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.8.1) 50 გ ცხოველის საკონტროლო საკვებში, კარგად აურიეთ და გააჩერეთ 10 წთ. ვიდრე გააგრძელებდეთ (ამ თავის პუნქტი 5.2) კვლავ რამდენჯერმე აურიეთ.
ალტერნატიულად, თუ ნიმუშის მსგავსი ტიპის ცხოველის საკონტროლო საკვები არ არის ხელმისაწვდომი (იხ. ამ თავის პუნქტი 5.1.1), აღდგენის ტესტი შეიძლება ჩატარდეს სტანდარტული დამატების მეთოდის საშუალებით. ამ შემთხვევაში, ხდება გამოსაკვლევი ნიმუშის გამდიდრება დიკლაზურილის იმ რაოდენობით, როგორც ეს უკვე წარმოდგენილია ნიმუშში. ხდება ამ ნიმუშის გამოკვლევა გაუმდიდრებელ ნიმუშთან ერთად და აღდგენის გამოანგარიშება შესაძლებელია სუბტრაქციის (გამოკლების) გზით.
5.2. ექსტრაჰირება 5.2.1. ცხოველის საკვები ნიმუშის დაახლოებით 50 გ აწონეთ 0,01 გ სიზუსტით. გადაიტანეთ 500 მლ კონუსურ კოლბაში, დაამატეთ 1,00 მლ შიდა სტანდარტული ხსნარი (ამ თავის პუნქტი 3.9.2), 200 მლ ექსტრაჰირების გამხსნელი (ამ თავის პუნქტი 3.12) და კოლბას დაახურეთ საცობი. ანჯღრიეთ ნარევი სადღვებზე (ამ თავის პუნქტი 4.1) მთელი ღამის განმავლობაში. დალექვას აცადეთ 10 წუთი. 20 მლ სუპერნატანტის ალიქვოტი გადაიტანეთ შესაფერის მინის ჭურჭელში და გააზავეთ 20 მლ წყლით. ეს ხსნარი გადაიტანეთ ექსტრაჰირების კარტრიჯზე (ამ თავის პუნქტი 3.11) და გაატარეთ ვაკუუმის გამოყენებით (ამ თავის პუნქტი 4.5). კარტრიჯი გარეცხეთ 25 მლ ექსტრაჰირების გამხსნელის ნარევითა (ამ თავის პუნქტი 3.12) და წყლით, 65 + 35 (V + V). მოაცილეთ შეგროვებული ფრაქციები და გახსენით ნაერთები 25 მლ ექსტრაჰირების გამხსნელისა (ამ თავის პუნქტი 3.12) და წყლის, 80 + 20 (V + V) ნარევით. აორთქლეთ ეს ფრაქცია მანამ, სანამ არ გამოშრება როტორული ამაორთქლებლის საშუალებით (ამ თავის პუნქტი 4.3) 60oC ტემპერატურაზე. ნაშთი (ნარჩენები) გახსენით 1,0 მლ DMF-ში (ამ თავის პუნქტი 3.6), დაამატეთ 1,5 მლ წყალი (ამ თავის პუნქტი 3.1) და აურიეთ. გაფილტრეთ მემბრანული ფილტრის საშუალებით (ამ თავის პუნქტი 4.4). გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.3).
5.2.2. პრემიქსები ნიმუშის დაახლოებით 1 გრ. აწონეთ 0,001 გ სიზუსტით. გადაიტანეთ 500 მლ კონუსურ კოლბაში, დაამატეთ 1,00 მლ შიდა სტანდარტული ხსნარი (ამ თავის პუნქტი 3.9.3), 200 მლ ექსტრაჰირების გამხსნელი (ამ თავის პუნქტი 3.12) და საცობი გაუკეთეთ კოლბას. შეანჯღრიეთ ნარევი მთელი დღის განმავლობაში სადღვებზე (ამ თავის პუნქტი 4.1). დალექვას დაელოდეთ 10 წუთი. 10 000/p მლ (p = დიკლაზურილის ნომინალური შემცველობა მგ/კგ) სუპერნატანტის ალიქვოტი გადაიტანეთ შესაფერისი ზომის მრგვალძირიან კოლბაში. აორთქლეთ გამოშრობამდე შემცირებული წნევის ქვეშ 60oC ტემპერატურაზე როტორული ამაორთქლებლის საშუალებით (ამ თავის პუნქტი 4.3). ნაშთი (ნარჩენები) ხელახლა გახსენით 10,0 მლ DMF-ში (ამ თავის პუნქტი 3.6), დაამატეთ 15,0 მლ წყალი (ამ თავის პუნქტი 3.1) და აურიეთ. გადადით HPLC-ის განსაზღვრაზე (ამ თავის პუნქტი 5.3).
5.3. HPLC-ის განსაზღვრა 5.3.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები, შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან:
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, რამდენჯერმე მოახდინეთ საკალიბრო ხსნარის (ამ თავის პუნქტი 3.10) ინჯექტირება, რომელიც შეიცავს 2 მკგ/მლ-ს, სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები და შენარჩუნების დრო.
5.3.2. საკალიბრო ხსნარი რამდენჯერმე შეიყვანეთ 20 მკლ საკალიბრო ხსნარი (ამ თავის პუნქტი 3.10) და განსაზღვრეთ დიკლაზურილის და შიდა სტანდარტის საშუალო პიკის სიმაღლე (ფართობი).
5.3.3. ნიმუშის ხსნარი რამდენჯერმე შეიყვანეთ 20 მკლ ნიმუშის ხსნარი (ამ თავის პუნქტი 5.2.1 ან 5.2.2) და განსაზღვრეთ დიკლაზურილის საშუალო პიკური სიმაღლე (ფართობი) და შიდა სტანდარტული პიკები.
6. შედეგების დაანგარიშება 6.1. ცხოველის საკვები დიკლაზურილის შემცველობა w (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: hd,s = დიკლაზურილის პიკური სიმაღლე (ფართობი) ნიმუშის ხსნარში (ამ თავის პუნქტი 5.2.1); hi,s = შიდა სტანდარტის პიკის სიმაღლე (ფართობი) ნიმუშის ხსნარში (ამ თავის პუნქტი 5.2.1); hd,c = დიკლაზურილის პიკური სიმაღლე (ფართობი) საკალიბრო ხსნარში (ამ თავის პუნქტი 3.10); hi,c = შიდა სტანდარტის პიკის სიმაღლე (ფართობი) საკალიბრო ხსნარში (ამ თავის პუნქტი 3.10); cd,c = დიკლაზურილის კონცენტრაცია საკალიბრო ხსნარში მკგ/მლ (ამ თავის პუნქტი 3.10); m = გამოსაკვლევი პორციის წონა გ-ში; V = ნიმუშის ექსტრაქტის მოცულობა ამ თავის 5.2.1 პუნქტის შესაბამისად (ანუ 2,5 მლ)
6.2. პრემიქსები დიკლაზურილის შემცველობა w (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: hd,c = დიკლაზურილის პიკური სიმაღლე (ფართობი) საკალიბრო ხსნარში (ამ თავის პუნქტი 3.10); hi,c = შიდა სტანდარტის პიკის სიმაღლე (ფართობი) საკალიბრო ხსნარში (ამ თავის პუნქტი 3.10); hd,s = დიკლაზურილის პიკური სიმაღლე (ფართობი) ნიმუშის ხსნარში (ამ თავის პუნქტი 5.2.2); hi,s = შიდა სტანდარტის პიკის სიმაღლე (ფართობი) ნიმუშის ხსნარში (ამ თავის პუნქტი 5.2.2); cd,c = დიკლაზურილის კონცენტრაცია საკალიბრო ხსნარში მკგ/მლ (ამ თავის პუნქტი 3.10); m = გამოსაკვლევი პორციის წონა გ-ში; V = ნიმუშის ექსტრაქტის მოცულობა ამ თავის 5.2.2-ე პუნქტის შესაბამისად (ანუ 25 მლ); p = დიკლაზურილის ნომინალური შემცველობა მგ/კგ-ზე პრემიქსში.
7. შედეგების ვალიდაცია
7.1. იდენტიფიკაცია გამოსაკვლევი ნივთიერების (ანალიტის) იდენტურობის დადასტურება შესაძლებელია თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც დარდება ნიმუშის ექსტრაქტის სპექტრები (ამ თავის პუნქტი 5.2.1 ან 5.2.2) და საკალიბრო ხსნარი (ამ თავის პუნქტი 3.10).
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი (ამ თავის პუნქტი 5.2.1 ან 5.2.2) გამდიდრებულია სათანადო რაოდენობის საკალიბრო ხსნარის დამატებით (ამ თავის პუნქტი 3.10). დიკლაზურილის დამატებული რაოდენობა უნდა იყოს დიკლაზურილის სავარაუდო რაოდენობის მსგავსი, რომელიც აღმოჩენილ იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ დიკლაზურილის პიკის სიმაღლე და შიდა სტანდარტული პიკი. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს დიკლაზურილის მწვერვალის ორიგინალი სიგანის ან არაგამდიდრებული ნიმუშის ექსტრაქტის შიდა სტანდარტული პიკის ± 10%-ის ფარგლებში.
7.1.2. გამოვლენა დიოდური მატრიცით შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით: ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბსორბციის ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, იდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცის გამოვლენისთვის ეს ჩვეულებრივ ±2 ნმ-ის ფარგლებშია; ბ) 230-დან 320 ნმ-მდე შუალედში ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100% ფარდობითი აბსორბციის დიაპაზონში. ეს კრიტერიუმი შესაბამისობაშია მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული გამოსაკვლევი ნივთიერების (ანალიტის) შთანთქმის 15%-ს; გ) 230-დან 320 ნმ-მდე შუალედში ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი შესაბამისობაშია მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15% -ს.
თუ რომელიმე ამ კრიტერიუმებიდან არ სრულდება (არ არის შესაბამისობაში), გამოსაკვლევი ნივთიერების (ანალიტის) არსებობა არ დადასტურდება.
7.2. განმეორებადობა ერთსა და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – 30%-ს უფრო მაღალ მნიშვნელობასთან შედარებით დიკლაზურილის შემცველობის 0,5 მგ/კგ-დან 2,5 მგ/კგ-მდე; – 0,75 მგ/კგ-ს დიკლაზურილის შემცველობაზე 2,5 მგ/კგ და 5 მგ/კგ შორის; – 15%-ს უფრო მაღალ მნიშვნელობასთან შედარებით 5 მგ/კგ-ზე მეტი დიკლაზურილის შემცველობისას.
7.3. აღდგენა გამდიდრებული საკონტროლო ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 80%.
8. ერთობლივი კვლევის შედეგები 11 ლაბორატორიის მიერ ჩატარდა ერთობლივი კვლევა, რა დროსაც გამოკვლეულ იქნა 5 ნიმუში. ეს ნიმუშები შედგებოდა ორი პრემიქსისაგან; ერთი შერეული იყო ორგანულ მატრიცასთან (O 100) და მეორე არაორგანულ მატრიცასთან (A 100). თეორიული შემცველობაა 100 მგ დიკლაზურილი კგ-ზე. ფრინველის სამი შერეული ცხოველის საკვები შეიქმნა 3 სხვადასხვა მწარმოებლის მიერ (NL) (L1/Z1/K1). თეორიული შემცველობა 1 მგ დიკლაზურილი კგ-ზე. ლაბორატორიებს დაევალათ თითოეული ნიმუშის გამოკვლევა ერთხელ ან განმეორებით (დუბლიკატი). (ამ ერთობლივი კვლევის შესახებ უფრო დეტალური ინფორმაცია შეგიძლიათ იხილოთ ჟურნალში AOAC International, ტომი 77, No 6, 1994, გვ. 1359-1361). შედეგები მოცემულია ცხრილში:
L = ლაბორატორიების რაოდენობა; n = ერთეულის მნიშვნელობის რაოდენობა; sr = განმეორებადობის სტანდარტული გადახრა; CVr = განმეორებადობის ვარიაციის კოეფიციენტი, %; SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი %.
9. დაკვირვება დიკლაზურილზე რეაქცია ადრე ნაჩვენები უნდა ყოფილიყო, როგორც ხაზოვანი გაზომილი კონცენტრაციების მთელ დიაპაზონში.
თავი VII ნატრიუმის ლასალოციდის განსაზღვრა
Streptomyces lasaliensis-ის მიერ წარმოებული პოლიეთერის მონოკარბოქსილის მჟავას ნატრიუმის მარილი
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ ნატრიუმის ლასალოციდის შემცველობა ცხოველის საკვებსა და პრემიქსებში. გამოვლენის ზღვარი არის 5 მგ/კგ, რაოდენობრივი შეფასების ზღვარი – 10 მგ/კგ.
2. გამოყენების პრინციპი ნატრიუმის ლასალოციდი ექსტრაჰირდება ნიმუშიდან მჟავაშეზავებულ მეთანოლში და განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (HPLC) სპექტროფლუორომეტრიული დეტექტორის გამოყენებით.
3. რეაგენტები 3.1. კალიუმის დიჰიდროფოსფატი (KH2 PO 4). 3.2. ორთოფოსფორის მჟავა, w (w/w) = 85%. 3.3. ორთოფოსფორის მჟავას ხსნარი, c = 20%. 23,5 მლ ორთოფოსფორის მჟავა (ამ თავის პუნქტი 3.2) 100 მლ-მდე განაზავეთ წყლით. 3.4. 6-მეთილ-2-ჰეპტილამინი (1,5-დიმეთილჰექსილამინი), w (w/w) = 99%. 3.5. მეთანოლი, HPLC ხარისხის ეკვივალენტი. 3.6. მარილმჟავა, სიმკვრივე = 1,19 გ/მლ. 3.7. ფოსფატის ბუფერული ხსნარი, c = 0,01 მოლი/ლიტრი. გახსენით 1,36 გ KH2 PO4 (ამ თავის პუნქტი 3.1) 500 მლ წყალში (ამ თავის პუნქტი 3,11), დაამატეთ 3,5 მლ ორთოფოსფორმჟავა (ამ თავის პუნქტი 3.2) და 10,0 მლ 6-მეთილ-2-ჰეპტილამინი (ამ თავის პუნქტი 3.4). pH მიიყვანეთ 4,0-მდე ორთოფოსფორმჟავის ხსნარით (ამ თავის პუნქტი 3.3) და წყლით განაზავეთ 1 000 მლ-მდე (ამ თავის პუნქტი 3.11). 3.8. მჟავაშერეული მეთანოლი. 5,0 მლ მარილმჟავა (ამ თავის პუნქტი 3.6) გადაიტანეთ 1 000 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე მეთანოლით (ამ თავის პუნქტი 3.5) და აურიეთ. გამოყენებამდე ეს ხსნარი უნდა იყოს ახალი მომზადებული. 3.9. HPLC მობილური ფაზა, ფოსფატის ბუფერულ-მეთანოლის ხსნარი 5 + 95 (V + V). შეურიეთ 5 მლ ფოსფატის ბუფერული ხსნარი (ამ თავის პუნქტი 3.7) 95 მლ მეთანოლთან (ამ თავის პუნქტი 3.5). 3.10. გარანტირებული სიწმინდის მქონე ნატრიუმის ლასალოციდის სტანდარტული ნივთიერება, C34H53O8Na (Streptomyces lasaliensis-ის მიერ წარმოებული პოლიეთერის მონოკარბოქსილის მჟავას ნატრიუმის მარილი), E763. 3.10.1. ნატრიუმის ლასალოციდის საწყისი სტანდარტული ხსნარი, 500 მკგ/მლ აწონეთ 0,1 მგ სიზუსტით, 50 მგ ნატრიუმის ლასალოციდის (ამ თავის პუნქტი 3,10) 100 მლ გრადირებულ კოლბაში, გახსენით მჟავაშეზავებულ მეთანოლში (ამ თავის პუნქტი 3,8), შეავსეთ ნიშნულამდე იმავე გამხსნელით და აურიეთ. გამოყენებამდე ეს ხსნარი უნდა იყოს ახალი მომზადებული.
3.10.2. ნატრიუმის ლასალოციდი შუალედური სტანდარტული ხსნარი, 50 მკგ/მლ 3.10.3. საკალიბრო ხსნარები 50 მლ გრადირებული კოლბების სერიაში გადაიტანეთ 1,0, 2,0, 4,0, 5,0 და 10,0 მლ შუალედური სტანდარტული ხსნარი (ამ თავის პუნქტი 3.10.2). შეავსეთ ნიშნულამდე მჟავაშეზავებული მეთანოლით (ამ თავის პუნქტი 3.8) და აურიეთ. ეს ხსნარები შეესაბამება 1,0, 2,0, 4,0, 5,0 და 10,0 მკგ ნატრიუმის ლასალოციდის მლ-ზე შესაბამისად. გამოყენებამდე ეს ხსნარები უნდა იყოს ახალი მომზადებული. 3.11. წყალი, HPLC ხარისხის ეკვივალენტი. 4. აპარატურა 4.1. ულტრაბგერითი აბაზანა (ან წყლის აბაზანა შეიკერით) ტემპერატურის კონტროლით. 4.2. მემბრანული ფილტრები, 0,45 მკმ. 4.3. HPLC მოწყობილობა ინჯექტირების სისტემით, შესაფერისია 20 მკლ მოცულობის ინჯექტირებისთვის 4.3.1. თხევადი ქრომატოგრაფიული სვეტი 125 მმ x 4 მმ, შებრუნებული ფაზის C18, 5 მკმ-იანი შეფუთვა ან ეკვივალენტი. 4.3.2. სპექტროფლუორომეტრი აგზნების და გამოსხივების ტალღის სიგრძის ცვალებადი ტალღის მარეგულირებელით.
5. გამოყენების პროცედურა 5.1. ზოგადი პრონციპები 5.1.1. ცხოველის საკონტროლო საკვები აღდგენითი ტესტის (ამ თავის პუნქტი 5.1.2) შესასრულებლად უნდა მოხდეს ცხოველის საკონტროლო საკვების გამოკვლევა, რათა გამოირიცხოს ნატრიუმის ლასალოციდისა და ხელშემშლელი ნივთიერებების არსებობა. ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის ტიპის მსგავსი და კვლევის დროს არ უნდა გამოვლინდეს ნატრიუმის ლაზალოციდი ან ხელშემშლელი ნივთიერებები.
5.1.2. აღდგენის ტესტი აღდგენის ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების კვლევით, რომელიც გამდიდრებულ იქნა ნატრიუმის ლასალოციდის იმ რაოდენობის დამატებით, როგორც ეს წარმოდგენილია ნიმუშში. 100 მგ/კგ დონეზე გამდიდრებისთვის დაამატეთ 10,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.10.1) გადაიტანეთ 250 მლ კონუსურ კოლბაში და ააორთქლეთ ხსნარი დაახლოებით 0,5 მლ-მდე. დაამატეთ 50 გ ცხოველის საკონტროლო საკვები, კარგად აურიეთ და გააჩერეთ 10 წუთი, კვლავ რამდენჯერმე აურიეთ, სანამ გადახვალთ ექსტრაჰირების ეტაპზე (ამ თავის პუნქტი 5.2).
ალტერნატიულად, თუ ნიმუშის მსგავსი ტიპის ცხოველის საკონტროლო საკვები არ არის ხელმისაწვდომი (იხ. ამ თავის პუნქტი 5.1.1), აღდგენის ტესტი შეიძლება ჩატარდეს სტანდარტული დამატების მეთოდის საშუალებით. ამ შემთხვევაში, ხდება გამოსაკვლევი ნიმუშის გამდიდრება ნატრიუმის ლასალოციდის რაოდენობით, ანალოგიურად, როგორც ეს უკვე წარმოდგენილია ნიმუშში. ხდება ამ ნიმუშის გამოკვლევა გაუმდიდრებელ ნიმუშთან ერთად და აღდგენის გამოანგარიშება შესაძლებელია სუბტრაქციის (გამოკლების) გზით.
5.2. ექსტრაჰირება 5.2.1. ცხოველის საკვები 5-დან 10გრ-მდე ნიმუში აწონეთ 0,01 გ-მდე სიზუსტით, საცობიან კონუსურ კოლბაში. პიპეტით დაამატეთ 100,0 მლ მჟავაშეზავებული მეთანოლი (ამ თავის პუნქტი 3.8). საცობს მოუჭირეთ და წრიულად აანჯღრიეთ შესარევად. კოლბა მოათავსეთ ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.1) დაახლოებით 40°C ტემპერატურაზე 20 წუთის განმავლობაში, შემდეგ ამოიღეთ და გააგრილეთ ოთახის ტემპერატურაზე. გააჩერეთ დაახლოებით 1 საათი, სანამ სუსპენზირებული ნივთიერება არ დაილექება, შემდეგ გაფილტრეთ ალიქვოტის პორცია 0.45 მკმ მემბრანული ფილტრის (ამ თავის პუნქტი 4.2) საშუალებით შესაბამის ჭურჭელში. გადადით HPLC-ის განსაზღვრაზე (ამ თავის პუნქტი 5.3).
5.2.2. პრემიქსები აწონეთ 0,001 გ-მდე სიზუსტით 2 გ დაუქუცმაცებელი პრემიქსის 250 მლ გრადირებულ (დანაყოფებიან) კოლბაში. დაამატეთ 100,0 მლ მჟავაშეზავებული მეთანოლი (ამ თავის პუნქტი 3.8) და წრიულად ატრიალეთ, რომ დაიშალოს. კოლბა და შემცველობა მოათავსეთ ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.1) დაახლოებით 40°C ტემპერატურაზე 20 წუთის განმავლობაში, შემდეგ ამოიღეთ და გააგრილეთ ოთახის ტემპერატურაზე. შეავსეთ ნიშნულამდე მჟავაშეზავებული მეთანოლით (ამ თავის პუნქტი 3.8) და კარგად აურიეთ. გააჩერეთ 1 საათით, სანამ სუსპენზირებული ნივთიერება არ დაილექება, შემდეგ გაფილტრეთ ალიქვოტის პორცია 0,45 მკმ მემბრანულ ფილტრში (ამ თავის პუნქტი 4.2). გასუფთავებული ფილტრატის შესაბამისი მოცულობა განაზავეთ მჟავაშერეული მეთანოლით (ამ თავის პუნქტი 3.8), რათა წარმოიქმნას საბოლოო გამოსაკვლევი ხსნარი, რომელიც შეიცავს ნატრიუმის ლასალოციდის დაახლოებით 4 მკგ/მლ. გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.3).
5.3. HPLC-ის განსაზღვრა
5.3.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან.
4,0 მკგ/მლ შემცველობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.10.3) რამდენჯერმე ინჯექტირების გზით, შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები (ან ფართობები) და შენარჩუნების დრო.
5.3.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის (ამ თავის პუნქტი 3.10.3) ინჯექტირება და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი, საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბცისასი.
5.3.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის ინჯექტირება (ამ თავის პუნქტი 5.2.1 ან 5.2.2), იმავე მოცულობის გამოყენებით, რაც აღებული იქნა საკალიბრო ხსნარებისთვის და განსაზღვრეთ ნატრიუმის ლასალოციდის სიმაღლეების საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების გაანგარიშება საშუალო პიკის სიმაღლიდან (ფართობიდან), რომელიც წარმოიქმნება ნიმუშის ხსნარის (ამ თავის პუნქტი 5.3.3) ინჯექტირების განსაზღვრეთ ნატრიუმის ლასალოციდის კონცენტრაცია (მკგ/მლ) საკალიბრო მრუდის მითითებით.
6.1. ცხოველის საკვები ნატრიუმის ლასალოციდის შემცველობა, w (მგ/კგ) ნიმუშში მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის ნატრიუმის ლასალოციდის კონცენტრაცია (ამ თავის პუნქტი 5.2.1) მკგ/მლ-ში; V1 = ნიმუშის ექსტრაქტის მოცულობა მლ-ში (მაგ. 100), ამ თავის 5.2.1-ე პუნქტის შესაბამისად; m = გამოსაკვლევი პორციის წონა გ-ში.
6.2. პრემიქსები ნატრიუმის ლასალოციდის შემცველობა, w (მგ/კგ) ნიმუშში მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის ნატრიუმის ლასალოციდის კონცენტრაცია (ამ თავის პუნქტი 5.2.2) მკგ/მლ-ში; V 2 = ნიმუშის ექსტრაქტის მოცულობა მლ-ში (მაგ. 250), ამ თავის 5.2.1-ე პუნქტის შესაბამისად; f = განზავების ფაქტორი ამ თავის 5.2.2-ე პუნქტის შესაბამისად; m = გამოსაკვლევი პორციის წონა გ-ში.
7. შედეგების ვალიდაცია 7.1. იდენტიფიკაცია სპექტროფლუორომეტრიაზე დაფუძნებული მეთოდები ნაკლებად ექვემდებარება ჩარევას, ვიდრე ის, რომლებშიც გამოიყენება UV გამოვლენა. გამოსაკვლევი ნივთიერების (ანალიტის) იდენტიფიკაცია შეიძლება დადასტურდეს თანაქრომატოგრაფიით.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი (ამ თავის პუნქტი 5.2.1 ან 5.2.2) გამდიდრებულია შესაბამისი რაოდენობის საკალიბრო ხსნარის დამატებით (ამ თავის პუნქტი 3.10.3). ლასალოციდის დამატებული რაოდენობა უნდა იყოს ნატრიუმის ლასალოციდის სავარაუდო რაოდენობის მსგავსი, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში. ნატრიუმის ლასალოციდის დამატებული ოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ, უნდა გაიზარდოს მხოლოდ ნატრიუმის ლასალოციდის პიკის სიმაღლე. მწვერვალის სიგანე, ნახევარ სიმაღლეზე, უნდა იყოს თავდაპირველი სიგანის ± 10%-ის ფარგლებში, რომელიც მიღებულია გაუმდიდრებელი ნიმუშის ექსტრაქტით.
7.2. განმეორებადობა ერთსა და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – 15%-ს უფრო მაღალ მნიშვნელობასთან შედარებით ნატრიუმის ლასალოციდის შემცველობის 30 მგ/კგ-დან 100 მგ/კგ-მდე; – 15 მგ/კგ-ს ნატრიუმის ლასალოციდის 100 მგ/კგ და 200 მგ/კგ შორის შემცველობისას; – 7,5%-ს უფრო მაღალ მნიშვნელობასთან შედარებით 200 მგ/კგ-ზე მეტი ნატრიუმის ლასალოციდის შემცველობისას.
7.3. აღდგენა გამდიდრებული (საკონტროლო) ცხოველის საკვების ნიმუშისთვის აღდგენა უნდა იყოს სულ მცირე 80%. გამდიდრებული პრემიქსის ნიმუშებისთვის აღდგენა უნდა იყოს სულ მცირე 90%.
8. ერთობლივი გამოკვლევის შედეგები ჩატარდა ერთობლივი კვლევა (*), რომელის დროსაც 12 ლაბორატორიამ გამოიკვლია 2 პრემიქსი (1 და 2 ნიმუში) და 5 ცხოველის საკვები (3-7 ნიმუშები). თითოეულ ნიმუშზე ჩატარდა განმეორებითი (დუბლიკატი) კვლევა. შედეგები მოცემულია მომდევნო ცხრილში:
L = ლაბორატორიების რაოდენობა; n = ერთეულის შედეგის რაოდენობა; sr = განმეორებადობის სტანდარტული გადახრა; SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; CVr = განმეორებადობის ვარიაციის კოეფიციენტი, %; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი %.
დანართი №5 ცხოველის საკვებში არასასურველი ნივთიერებების კონტროლისათვის გამოკვლევის მეთოდები
თავი I თავისუფალი გოსიპოლის და გოსიპოლის საერთო რაოდენობის განსაზღვრა 1. მიზანი და გამოყენების სფერო ეს მეთოდი განსაზღვრავს თავისუფალი გოსიპოლის, გოსიპოლის საერთო რაოდენობისა და მასთან ქიმიურად დაკავშირებული ნივთიერებების შემცველობას ბამბის თესლში, ბამბის თესლის მსხვილად ნაფქვავისა და ბამბის თესლის ნაწნეხი და ამ მასალის შემცველი ცხოველის კომბინირებულ საკვებში, როდესაც თავისუფალი გოსიპოლი, გოსპიპოლის საერთო შემცველობა და მასთან ქიმიურად დაკავშირებული ნივთიერებები 20მგ/კგ-ზე მეტი კონცენტრაციით არის წარმოდგენილი. 2. გამოყენების პრინციპი გოსიპოლის მოპოვება ხდება 3-ამინოპროპან-1-ოლის, ან პროპან-2-ოლის და ჰექსანის ნარევთან ერთად, თავისუფალი გოსიპოლის დასადგენად, ან დიმეთილფორმამიდით, გოსიპოლის საერთო რაოდენობის დასადგენად. გოსიპოლი ანილინით გარდაიქმნება გოსიპოლ-დიანილინად, რომლის ოპტიკური სიმკვრივე იზომება 440 ნმ-ზე. 3. რეაგენტები 3.1. პროპან-2-ოლ-ჰექსანის ნარევი: პროპან-2-ოლის მოცულობის 60 ნაწილი აურიეთ N – ჰექსანის 40 მოცულობით ნაწილთან. 3.2. გამხსნელი A: მოათავსეთ 1-ლიტრიან გრადირებულ კოლბაში დაახლოებით 500 მლ პროპან-2-ოლ-ჰექსანის ნარევი (ამ თავის პუნქტი 3.1), 2 მლ 3-ამინოპროპან-1-ოლი, 8 მლ დაკრისტალებული ძმარმჟავა და 50 მლ წყალი. შეავსეთ მოცულობა პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1). ეს რეაგენტი სტაბილურია ერთი კვირის განმავლობაში. 3.3. გამხსნელი B: 2 მლ 3-ამინოპროპან-1-ოლი და 10 მლ დაკრისტალებული ძმარმჟავა 100 მლ პიპეტით გადაიტანეთ გრადირებულ კოლბაში. გააგრილეთ ოთახის ტემპერატურაზე და შეავსეთ მოცულობა N, N – დიმეთილფორმამიდით. ეს რეაგენტი სტაბილურია ერთი კვირის განმავლობაში. 3.4. ანილინი: თუ საკონტროლო ტესტის ოპტიკური სიმკვრივე აღემატება 0,022-ს, ანილინი გამოხადეთ თუთიის მტვერზე, ამასთან მოაცილეთ დისტილატის პირველი და ბოლო ფრაქციების 10%. შედგით მაცივარში და შეინახეთ ყავისფერ, დახურულ მინის კოლბაში, ეს რეაგენტი რამდენიმე თვის განმავლობაში შენარჩუნდება. 3.5. გოსიპოლის სტანდარტული ხსნარი A: მოათავსეთ 27,9 მგ გოსიპოლის აცეტატი 250 მლ გრადირებულ კოლბაში. გახსენით და შეავსეთ მოცულობა A გამხსნელით (ამ თავის პუნქტი 3.2). ამ ხსნარის 50 მლ გადაიტანეთ პიპეტით 250 მლ გრადუსულ კოლბაში და შეავსეთ მოცულობა A გამხსნელით. ამ ხსნარის გოსიპოლის კონცენტრაცია არის 0,02 მგ/მლ. გამოყენებამდე გააჩერეთ ერთი საათის განმავლობაში ოთახის ტემპერატურაზე. 3.6. გოსიპოლის სტანდარტული ხსნარი B: 27,9 მგ გოსიპოლის აცეტატი მოათავსეთ 50 მლ გრადირებულ კოლბაში, გახსენით და შეავსეთ მოცულობა B გამხსნელით (ამ თავის პუნქტი 3.3). ამ ხსნარის გოსიპოლის კონცენტრაცია არის 0,5 მგ/მლ. გოსიპოლის სტანდარტული A და B ხსნარები სტაბილურია 24 საათის განმავლობაში, თუ დაცული იქნება სინათლისგან. 4. აპარატურა 4.1. მიქსერი (ტუმბლერი): დაახლოებით 35 ბრ./წ.თ. 4.2. სპექტროფოტომეტრი. 5. გამოყენების პროცედურა 5.1 გამოსაკვლევი ნიმუში გამოყენებული გამოსაკვლევი ნიმუშის რაოდენობა დამოკიდებულია ნიმუშში გოსიპოლის სავარაუდო შემცველობაზე. სასურველია მუშაობა მცირე გამოსაკვლევ ნიმუშთან და ფილტრატის ალიქვოტის შედარებით დიდ პორციასთან, რათა მივიღოთ საკმარისი გოსიპოლი ზუსტი ფოტომეტრიული გაზომვისთვის. ბამბის თესლში, ბამბის თესლის მსხვილად ნაფქვავისა და ბამბის თესლის ნაწნეხში თავისუფალი გოსიპოლის დასადგენად, ნიმუში არ უნდა აღემატებოდეს 1 გ-ს; ცხოველის კომბინირებული საკვებისთვის შეიძლება იყოს 5 გრამი. 10 მლ ფილტრატის ალიქვოტის ნაწილი საკმარისია უმეტესი შემთხვევებისთვის; იგი უნდა შეიცავდეს 50-დან 100 მკგ გოსიპოლს. გოსიპოლის საერთო რაოდენობის დასადგენად, გამოსაკვლევი ნიმუში უნდა იყოს 0,5-დან 5 გ-მდე, იმისათვის რომ ფილტრატის 2 მლ ალიქვოტის პორცია შეიცავდეს 40-დან 200 მკგ გოსიპოლს. გამოკვლევა უნდა ჩატარდეს ოთახის ტემპერატურაზე, დაახლოებით 20oC-ზე.
5.2. თავისუფალი გოსიპოლის განსაზღვრა გამოსაკვლევი ნიმუში მოათავსეთ გლუვყელიან 250-მლ-იან კოლბაში, კოლბის ფსკერი დაფარული უნდა იყოს დაქუცმაცებული მინით. პიპეტის გამოყენებით დაამატეთ 50 მლ A გამხსნელი (ამ თავის პუნქტი 3.2), კოლბას გაუკეთეთ საცობი და მიქსერში ურიეთ ერთი საათის განმავლობაში. გაფილტრეთ მშრალი ფილტრის საშუალებით და შეაგროვეთ ფილტრატი მცირე ზომის გლუვყელიან კოლბაში. ფილტრაციის დროს ძაბრს (ხვრელს) დააფარეთ საათის მინა ჭიქა. ფილტრატის იდენტური ალიქვოტის პორცია, რომელიც შეიცავს 50-დან 100 მკგ-მდე გოსიპოლს, პიპეტით ცალ-ცალკე გადაიტანეთ ორ 25-მლ-იან გრადირებულ კოლბაში (A და B). საჭიროების შემთხვევაში, მოცულობა შეავსეთ 10 მლ-მდე A გამხსნელით (ამ თავის პუნქტი 3.2). შემდეგ, კოლბის (A) შემცველობა შეავსეთ პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1). ეს ხსნარი გამოყენებული იქნება, როგორც ეტალონური ხსნარი, რომლის მიმართაც უნდა მოხდეს ნიმუშის ხსნარის გაზომვა. პიპეტით გადაიტანეთ 10 მლ გამხსნელი A (ამ თავის პუნქტი 3.2) ცალ-ცალკე ორ 25-მლ-იან გრადირებულ კოლბაში (C და D). კოლბის (C) შემცველობა შეავსეთ მოცულობამდე პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1). ეს ხსნარი გამოყენებული უნდა იქნეს, როგორც ეტალონური ხსნარი, რომლის მიმართაც უნდა მოხდეს საკონტროლო ტესტის ხსნარის გაზომვა. თითოეულ (D) და (B) კოლბაში დაამატეთ 2 მლ ანილინი (ამ თავის პუნქტი 3.4). გააცხელეთ 30 წუთის განმავლობაში მდუღარე წყლის აბაზანაზე, ფერის მისაღებად. გააგრილეთ ოთახის ტემპერატურამდე, შეავსეთ მოცულობა პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1), მოახდინეთ ჰომოგენიზება და სტაბილიზაციისთვის გააჩერეთ ერთი საათის განმავლობაში. განსაზღვრეთ საკონტროლო ტესტის ხსნარის (D) ოპტიკური სიმკვრივე, ეტალონურ ხსნართან (C) შედარების გზით, ხოლო საკონტროლო ტესტის ხსნარის (B) ოპტიკური სიმკვრივე, ეტალონურ ხსნართან (A) შედარების გზით, სპექტროფოტომეტრში 440 ნმ-ზე 1 სმ-იანი მინის უჯრედების გამოყენებით. მოახდინეთ საკონტროლო ტესტის ხსნარის ოპტიკური სიმკვრივის ნიმუშის ხსნარიდან გამოკლება (= შესწორებული ოპტიკური სიმკვრივე). ამ მნიშვნელობიდან გამოთვალეთ თავისუფალი გოსიპოლის შემცველობა, როგორც მითითებულია ამ თავის მე-6 პუნქტში. 5.3. გოსიპოლის საერთო რაოდენობის განსაზღვრა მოათავსეთ გამოსაკვლევი ნიმუში, რომელიც შეიცავს 1-დან 5 მგ გოსიპოლს, 50 მლ გრადირებულ კოლბაში და დაამატეთ 10 მლ გამხსნელი B (ამ თავის პუნქტი 3.3). ამავდროულად, მოამზადეთ საკონტროლო ტესტი, 10 მლ B გამხსნელის (ამ თავის პუნქტი 3.3) სხვა 50 მლ გრადირებულ კოლბაში მოთავსებით. გაათბეთ ორივე კოლბა 30 წუთის განმავლობაში მდუღარე წყლის აბაზანაზე. გააგრილეთ ოთახის ტემპერატურამდე, შეავსეთ თითოეული კოლბის მოცულობა პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1). მოახდინეთ ჰომოგენიზება და გააჩერეთ 10-დან 15 წუთის განმავლობაში, შემდეგ გაფილტრეთ და შეაგროვეთ ფილტრები გლუვყელიან კოლბებში. 2 მლ ნიმუშის ფილტრატი პიპეტით გადაიტანეთ ცალ-ცალკე ორ 25-მლ-იან გრადირებულ კოლბაში, ხოლო 2 მლ საკონტროლო ტესტის ფილტრატი ცალ-ცალკე ორ 25-მლ-იან სხვა კოლბაში. თითოეული სერიიდან ერთი კოლბის შემცველობა შეავსეთ 25 მლ-მდე პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1). ეს ხსნარები გამოყენებული უნდა იქნეს, როგორც ეტალონური ხსნარები. დანარჩენ ორ კოლბს დაუმატეთ 2 მლ ანილინი (ამ თავის პუნქტი 3.4). გააცხელეთ 30 წუთის განმავლობაში მდუღარე წყლის აბაზანაში, ფერის მისაღებად. გააგრილეთ ოთახის ტემპერატურამდე, შეავსეთ 25 მლ-მდე პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1), მოახდინეთ ჰომოგენიზება, და გააჩერეთ ერთი საათის განმავლობაში. განსაზღვრეთ ოპტიკური სიმკვრივე, როგორც ეს ნაჩვენებია ამ თავის 5.2-ე პუნქტში თავისუფალი გოსიპოლისთვის. ამ მნიშვნელობიდან გამოთვალეთ გოსიპოლის საერთო შემცველობა, როგორც მითითებულია ამ თვაის მე-6 პუნქტში.
6. შედეგების დაანგარიშება შედეგები გამოითვლება, სპეციფიკური ოპტიკური სიმკვრივისგან (ამ თავის პუნქტი 6.1), ან საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 6.2). 6.1. სპეციფიკური ოპტიკური სიმკვრივისგან სპეციფიკური ოპტიკური სიმკვრივე, აღწერილ პირობებში, იქნება შემდეგი:
თავისუფალი გოსიპოლი:
გოსიპოლის საერთო რაოდენობა: ნიმუშში თავისუფალი გოსიპოლის ან გოსიპოლის საერთო რაოდენობის შემცველობა გამოითვლება შემდეგი ფორმულის გამოყენებით:
სადაც: E = შესწორებული ოპტიკური სიმკვრივე, რომელიც განისაზღვრება, როგორც ეს მითითებულია ამ თავის 5.2-ე პუნქტში, p = გამოსაკვლევი ნიმუში გრამებში, a = ფილტრატის ალიქვოტის პორცია მლ-ში. 6.2. საკალიბრო მრუდის გამოყენებით 6.2.1. თავისუფალი გოსიპოლი მოამზადეთ ორი სერია, ხუთ-ხუთი 25 მლ გრადირებული კოლბა. 2,0, 4,0, 6,0, 8,0 და 10,0 მლ ალიქვოტები სტანდარტული გოსიპოლის ხსნარი A (ამ თავის პუნქტი 3.5), პიპეტით გადაიტანეთ კოლბების თითოეულ სერიაში. შეავსეთ მოცულობა 10 მლ-მდე A გამხსნელით (ამ თავის პუნქტი 3.2). თითოეული სერია შეავსეთ 25 მლ გრადირებული კოლბით, რომელიც შეიცავს მხოლოდ 10 მლ გამხსნელს A (ამ თავის პუნქტი 3.2) (საკონტროლო ტესტი). პირველი სერიის კოლბების მოცულობა (საკონტროლო ტესტის კოლბის ჩათვლით) 25 მლ-მდე შეავსეთ პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1) (რეფერენს სერია). მეორე სერიის თითოეულ კოლბას დაუმატეთ 2 მლ ანილინი (ამ თავის პუნქტი 3.4) (საკონტროლო ტესტის კოლბის ჩათვლით). გააცხელეთ 30 წუთის განმავლობაში მდუღარე წყლის აბაზანაში, ფერის მისაღებად. გააგრილეთ ოთახის ტემპერატურამდე, შეავსეთ მოცულობა პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1), მოახდინეთ ჰომოგენიზება და გააჩერეთ ერთი საათის განმავლობაში (სტანდარტული სერია). ამ თავის 5.2-ე პუნქტში აღწერილი მითითებების მიხედვით, განსაზღვრეთ სტანდარტული სერიების ხსნარების ოპტიკური სიმკვრივე ეტალონური სერიის შესაბამის ხსნარებთან შედარების გზით. მოახდინეთ საკალიბრო მრუდის მიკვლევა, გოსიპოლის რაოდენობების მიმართ ოპტიკური სიმკვრივის დადგენით (მკგ-ში). 6.2.2. გოსიპოლის საერთო რაოდენობა მოამზადეთ ექვსი 50-მლ-იანი გრადირებული კოლბა. პირველ კოლბაში მოათავსეთ 10 მლ გამხსნელი B (ამ თავის პუნქტი 3.3), დანარჩენებში კი შესაბამისად, 2,0, 4,0, 6,0, 8,0 და 10,0 მლ სტანდარტული გოსიპოლის ხსნარი B (ამ თავის პუნქტი 3.6). თითოეული კოლბის შემცველობა 10 მლ-მდე შეავსეთ B გამხსნელით (ამ თავის პუნქტი 3.3). გაათბეთ 30 წუთის განმავლობაში მდუღარე წყლის აბაზანაში. გააგრილეთ ოთახის ტემპერატურამდე, შეავსეთ მოცულობა პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1) და მოახდინეთ ჰომოგენიზება. ამ ხსნარების 2,0 მლ მოათავსეთ თითოეულ ამ ორი სერიის ექვს 25-მლ-იან გრადირებულ კოლბაში. პირველი სერიის კოლბების შემცველობა შეავსეთ 25 მლ-მდე პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1) (რეფერენს სერია). მეორე სერიის თითოეულ კოლბაში დაამატეთ 2 მლ ანილინი (ამ თავის პუნქტი 3.4). გაათბეთ 30 წუთის განმავლობაში მდუღარე წყლის აბაზანაში. გააგრილეთ ოთახის ტემპერატურამდე, შეავსეთ მოცულობა პროპან-2-ოლ-ჰექსანის ნარევით (ამ თავის პუნქტი 3.1), მოახდინეთ ჰომოგენიზება და გააჩერეთ ერთი საათის განმავლობაში (სტანდარტული სერია). ამ თავის 5.2-ე პუნქტში აღწერილი მითითებების მიხედვით, განსაზღვრეთ სტანდარტული სერიების ხსნარების ოპტიკური სიმკვრივე რეფერენს სერიის შესაბამის ხსნარებთან შედარების გზით. მოახდინეთ საკალიბრო მრუდის მიკვლევა, გოსიპოლის რაოდენობების მიმართ ოპტიკური სიმკვრივის დადგენით (მკგ-ში). 6.3. განმეორებადობა ერთი და იმავე ნიმუშზე განხორციელებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – 15%-ს, უმაღლეს მნიშვნელობასთან შედარებისას, გოსიპოლის 500 ppm-ზე ნაკლები შემცველობისთვის, – 75 ppm-ს, აბსოლუტურ მნიშვნელობაში, გოსიპოლის არანაკლებ 500 ppm და არაუმეტეს 750 ppm შემცველობისთვის, – 10%-ს, უმაღლეს მნიშვნელობასთან შედარებისას, გოსიპოლის 750 ppm-ზე მეტი შემცველობისთვის.
თავი II დიოქსინების (PCDD/PCDF) და PCB-ების შემცველობის განსაზღვრა ქვეთავი I ნიმუშის აღების მეთოდები და გამოკვლევის შედეგების ინტერპრეტაცია 1. მოქმედების სფერო და განმარტებები ის ნიმუშები, რომლებიც გათვალისწინებულია ცხოველის საკვებში პოლიქლორირებული დიბენზო-p-დიოქსინების (PCDD), პოლიქლორირებული დიბენზოფურანების (PCDF), დიოქსინის მსგავსი პოლიქლორირებული ბიფენილების (PCB)10 და არა დიოქსინის მსგავსი PCB-ის შემცველობის სახელმწიფო კონტროლისთვის, აღებული უნდა იქნეს ამ წესის დანართის №1 მოთხოვნათა შესაბამისად. რაოდენობრივი მოთხოვნები ნივთიერებების ან პროდუქტების კონტროლთან დაკავშირებით, რომლებიც ერთნაირად ნაწილდება ცხოველის საკვებში, როგორც ეს ამ ქვეთავის 5.1-ე პუნქტით არის გათვალისწინებული, გამოყენებული უნდა იქნეს ამ წესის დანართის №1. ამგვარად მიღებული გაერთიანებული ნიმუშები უნდა ჩაითვალოს რეპრეზენტატულ (წარმომადგენლობით) ნიმუშად იმ ლოტების ან ქველოტებისთვის, საიდანაც ისინი იქნა აღებული. საქართველოს კანონმდებლობით დადგენილი მაქსიმალური შემცველობის შესაბამისობა დგინდება ლაბორატორიულ ნიმუშებში განსაზღვრული შემცველობის საფუძველზე. ამ ქვეთავის მიზნებისათვის გამოიყენება განმარტებები, რომლებიც მოცემულია „ტექნიკური რეგლამენტის – ცოცხალ ცხოველებსა და ცხოველური წარმოშობის სურსათში ზოგიერთი ნივთიერებისა (სუბსტანციის) და მათი ნარჩენების გამოკვლევისათვის ანალიზის მეთოდების განხორციელებისა და შედეგების ინტერპრეტაციის წესის დამტკიცების შესახებ“ – საქართველოს მთავრობის 2016 წლის 8 ნოემბრის N499 დადგენილებაში. ამ განმარტებების გარდა ამ თავის მიზნებისათვის გამოიყენება შემდეგი განმარტებები: სკრინინგის მეთოდები – მეთოდები, რომლებიც გამოიყენება იმ ნიმუშების შესარჩევად, სადაც PCDD/F და დიოქსინის მსგავსი PCB-ის შემცველობა აღემატება მაქსიმალურ შემცველობას ან მოქმედების ზღურბლს. მათ უნდა უზრუნველყონ ნიმუშების ხარჯეფექტური მაღალი გამტარუნარიანობა, რაც გაზრდის მაღალი ექსპოზიციის ახალი ინციდენტებისა და მომხმარებელთათვის ჯანმრთელობის რისკების გამოვლენის შანსს. სკრინინგის მეთოდები უნდა ემყარებოდეს ბიოანალიტიკურ ან GC-MS მეთოდებს. მაქსიმალური შემცველობის შესაბამისობის შესამოწმებლად ზღვრულ მნიშვნელობაზე (cut-off value) გადაჭარბებული შედეგის მქონე ნიმუშების გადამოწმება (ვერიფიკაცია) უნდა მოხდეს ორიგინალი (თავდაპირველი) ნიმუშის სრული განმეორებითი გამოკვლევით, დამადასტურებელი მეთოდის გამოყენებით. დამადასტურებელი მეთოდები – მეთოდები, რომლებიც უზრუნველყოფენ სრულ ან დამატებით ინფორმაციას, რომლითაც შესაძლებელი იქნება PCDD/F და მსგავსი PCB-ების დადგენა და არაორაზროვანი რაოდენობრივი განსაზღვრა მაქსიმალურ, ან საჭიროების შემთხვევაში, მოქმედების ზღურბლზე. ასეთი მეთოდები იყენებენ გაზის ქრომატოგრაფიას/მაღალი გარჩევადობის მას-სპექტრომეტრიას (GC-HRMS), ან გაზის ქრომატოგრაფიას/ტანდემურ მას-სპექტრომეტრიას (GC-MS/MS). შენიშვნა: ცხრილი სქოლიო10-დან
2. ლოტის ან ქველოტის შესაბამისობა მაქსიმალურ შემცველობასთან (ზღვართან) 2.1. არადიოქსინის მსგავს PCB-ებთან დაკავშირებით ლოტი ან ქველოტი შეესაბამება მაქსიმალურ შემცველობას (ზღვარს), თუ გამოკვლევის შედეგი PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 და PCB 180 (შემდგომში არადიოქსინის მსგავსი PCB-ები) ჯამისთვის, რომელიც, განუსაზღვრელობის გაფართოებული განსაზღვრის11 გათვალისწინებით, არ აღემატება საქართველოს კანონმდებლობით განსაზღვრულ მაქსიმალურ შემცველობას (ზღვარს). ლოტი ან ქველოტი არ შეესაბამება საქართველოს კანონმდებლობით დადგენილ მაქსიმალურ შემცველობას (ზღვარს), თუ განმეორებითი (დუბლიკატი) გამოკვლევის12 შედეგად მიღებული ორი ზედა ზღვარის13 საშუალო, განუსაზღვრელობის გაფართოებული განსაზღვრის გათვალისწინებით, აღემატება მაქსიმალურ შემცველობას (ზღვარს) გონივრული ეჭვს მიღმა, ანუ გამოკვლეული კონცენტრაცია გამოიყენება შესაბამისობის შესაფასებლად განუსაზღვრელობის გაფართოებული განსაზღვრის გამორიცხვის შემდეგ. განუსაზღვრელობის გაფართოებული განსაზღვრა გამოითვლება დაფარვის ფაქტორი 2-ით, რომელიც იძლევა დაახლოებით 95%-ს სანდოობას. ლოტი ან ქველოტი შეუსაბამოა, თუ გაზომილი მნიშვნელობის საშუალო, გაფართოებული განუსაზღვრელობის საშუალოზე გამოკლებული, მეტია მაქსიმალურ შემცველობაზე (ზღვარზე). ამ პუნქტის ზემოთ მოცემული პუნქტით განსაზღვრული წესები გამოიყენება სახელმწიფო კონტროლის ნიმუშზე მიღებული გამოკვლევის შედეგისთვის. დაცვის ან რეფერენტული გამოკვლევის შემთხვევაში, სხვა წესების არარსებობის შემთხვევაშიც გამოიყენება ამ პუნქტის ზემოთ მოცემული პუნქტით განსაზღვრული წესები. 2.2. PCDD/F და დიოქსინის მსგავს PCB-თან დაკავშირებით ლოტი ან ქველოტი შეესაბამება მაქსიმალურ შემცველობას (ზღვარს), თუ ერთი გამოკვლევის შედეგი: – შესრულებულია სკრინინგის მეთოდით, 5%-ზე ნაკლები მცდარი შესაბამისობის კოეფიციენტით, რაც მიუთითებს იმაზე, რომ ეს ზღვარი არ აღემატება PCDD/F-ების შესაბამის მაქსიმალურ შემცველობას და PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამს, რომელიც დადგენილია საქართველოს კანონმდებლობით; – შესრულებულია დამადასტურებელი მეთოდით, არ აღემატება PCDD/F-ების შესაბამის მაქსიმალურ შემცველობას (ზღვარს) და PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამს, რომელიც დადგენილია საქართველოს კანონმდებლობით, განუსაზღვრელობის გაფართოებული განსაზღვრის გათვალისწინებით. სკრინინგის შეფასებისთვის უნდა განისაზღვროს ზღვრული მნიშვნელობა (cut-off value) იმის დასადგენად, შეესაბამება თუ არა ნიმუში შესაბამის მაქსიმალურ შემცველობას (ზღვარს) PCDD/F-ებისთვის ან PCDD/F და დიოქსინის მსგავსი PCB-ების ჯამისთვის. ლოტი ან ქველოტი არ შეესაბამება საქართველოს კანონმდებლობით განსაზღვრულ მაქსიმალურ შემცველობას (ზღვარს), თუ განმეორებითი (დუბლიკატი) გამოკვლევის14 შედეგად მიღებული ორი ზედა ზღვრის15 გამოკვლევის შედეგების საშუალო, განუსაზღვრელობის გაფართოებული განსაზღვრის გათვალისწინებით, აღემატება მაქსიმალურ შემცველობას (ზღვარს) გონივრული ეჭვს მიღმა, ანუ გამოკვლეული კონცენტრაცია გამოიყენება შესაბამისობის შესაფასებლად, განუსაზღვრელობის გაფართოებული განსაზღვრის გამორიცხვის შემდეგ. განუსაზღვრელობის გაფართოებული განსაზღვრა გამოითვლება დაფარვის ფაქტორი 2-ით, რომელიც იძლევა დაახლოებით 95%-ს სანდოობის დონეს. ლოტი ან ქველოტი შეუსაბამოა, თუ გაზომილი მნიშვნელობის საშუალო, გაფართოებული განუსაზღვრელობის საშუალოზე გამოკლებული, მეტია მაქსიმალურ შემცველობაზე (ზღვარზე). PCDD/F და დიოქსინის მსგავსი PCB-ების ცალკეული გამოკვლევის შედეგების სავარაუდო გაფართოებული განუსაზღვრელობის ჯამი გამოყენებული უნდა იქნეს PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამისთვის. ამ პუნქტის ზემოთ მოცემული პუნქტით განსაზღვრული წესები გამოიყენება სახელმწიფო კონტროლის ნიმუშზე მიღებული გამოკვლევის შედეგისთვის. დაცვის ან რეფერენტული გამოკვლევის შემთხვევაში, სხვა წესების არარსებობის შემთხვევაშიც, გამოიყენება ამ პუნქტის ზემოთ მოცემული პუნქტით განსაზღვრული წესები. 3. დიოქსინისა და დიოქსინის მსგავსი PCB-თვის საქართველოს კანონმდებლობით განსაზღვრული მოქმედების ზღურბლს გადაჭარბებული შედეგები მოქმედების ზღურბლი წარმოადგენს ნიმუშების შერჩევის ინსტრუმენტს იმ შემთხვევებში, როდესაც საჭიროა დაბინძურების წყაროს დადგენა და მისი შემცირების ან აღმოფხვრის მიზნით ღონისძიებების გატარება. სკრინინგის მეთოდებით უნდა განისაზღვროს შესაბამისი ზღვრული მნიშვნელობები (cut-off values) ამ ნიმუშების შერჩევისთვის. სადაც მნიშვნელოვანი ძალისხმევაა საჭირო წყაროს დასადგენად და დაბინძურების შესამცირებლად ან აღმოსაფხვრელად, მიზანშეწონილია მოქმედების ზღურბლების გადაცდენის დადასტურება განმეორებითი (დუბლიკატი) გამოკვლევით, რომელიც იყენებს დამადასტურებელ მეთოდს და რომელიც ითვალისწინებს განუსაზღვრელობის გაფართოებულ განსაზღვრას 16. ქვეთავი II ცხოველის საკვებში დიოქსინების (PCDD/Fs) და დიოქსინის მსგავსი PCB შემცველობის სახელმწიფო კონტროლისთვის ნიმუშის მომზადებასა და გამოკვლევის მეთოდებთან დაკავშირებული მოთხოვნები 1. გამოყენების სფერო ამ თავით განსაზღვრული მოთხოვნები გამოყენებული უნდა იქნეს სახელმწიფო კონტროლისთვის, როდესაც ცხოველის საკვები გამოკვლეულია 2,3,7,8-ჩანაცვლებული PCDD/F და დიოქსინის მსგავსი PCB შემცველობაზე და ნიმუშის მომზადებისა და გამოკვლევის მოთხოვნებთან დაკავშირებით სხვა მარეგულირებელი მიზნებისთვის, რომელიც მოიცავს ცხოველების საკვების ბიზნესოპერატორის მიერ განხორციელებულ კონტროლს, „ცხოველის საკვების ჰიგიენის წესის დამტკიცების შესახებ“ – საქართველოს მთავრობის 2020 წლის 13 მარტის N173 დადგენილებით განსაზღვრული მოთხოვნების შესაბამისობის უზრუნველსაყოფად. ცხოველის საკვებში PCDD/F და დიოქსინის მსგავსი PCB-ების არსებობის მონიტორინგი შეიძლება ჩატარდეს ორი განსხვავებული ტიპის გამოკვლევის მეთოდით: ა) სკრინინგის მეთოდები სკრინინგის მეთოდების მიზანია შეარჩიოს ის ნიმუშები, რომლებსაც აქვთ PCDD/F და დიოქსინის მსგავსი PCB, რომლებიც აღემატება მაქსიმალურ შემცველობას (ზღვარს) ან მოქმედების ზღურბლებს. სკრინინგის მეთოდებმა უნდა უზრუნველყოს ნიმუშების ხარჯეფექტური მაღალი გამტარუნარიანობა, რაც გაზრდის მომხმარებელთა მაღალი ექსპოზიციის და ჯანმრთელობის რისკებთან დაკავშირებული ახალი ინციდენტების აღმოჩენის შანსს. მათი გამოყენება მიზნად ისახავს ცრუ შესაბამისობის შედეგების თავიდან აცილებას. ისინი შეიძლება მოიცავდეს ბიოანალიტიკურ და GC-MS მეთოდებს. სკრინინგის მეთოდები ადარებს კვლევის შედეგს ზღვრულ მნიშვნელობასთან (cut-off value) იმ პირობით, რომ უზრუნველყოფს კი/არა-გადაწყვეტილებას მაქსიმალური შემცველობის (ზღვარის) ან მოქმედების ზღურბლის შესაძლო გადაჭარბებით. PCDD/F-ების კონცენტრაცია და PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამი იმ ნიმუშებში, რომლებიც სავარაუდოდ არ შეესაბამება მაქსიმალურ შემცველობას (ზღვარს), განისაზღვრება ან დასტურდება დამადასტურებელი მეთოდით. გარდა ამისა, სკრინინგის მეთოდებმა შეიძლება მიუთითონ PCDD/F და დიოქსინის მსგავსი PCB შემცველობის შესახებ ნიმუშში. ბიოანალიტიკური სკრინინგის მეთოდების გამოყენების შემთხვევაში, შედეგი გამოიხატება, როგორც ბიოანალიტიკური ეკვივალენტები (BEQ), ხოლო ფიზიკურ-ქიმიური GC-MS მეთოდების გამოყენების შემთხვევაში ის გამოიხატება, როგორც ტოქსიკური ეკვივალენტები (TEQ). სკრინინგის მეთოდების რიცხობრივად მითითებული შედეგები შესაფერისია მოქმედების ზღუბლების შესაბამისობის ან საეჭვო შეუსაბამობის ან გადაჭარბების დემონსტრირებისთვის და დამადასტურებელი მეთოდებით გამოყენების შემთხვევაში მიუთითებს შემცველობის დიაპაზონის შესახებ. ისინი არ არის შესაფერისი ისეთი მიზნებისთვის, როგორიცაა ფონის დონეების შეფასება, მიღების შეფასება, დონის დროის ტენდენციების დაცვა ან მოქმედების ზღუბლებისა და მაქსიმალური შემცველობის (ზღვარის) ხელახალი შეფასება. ბ) დამადასტურებელი მეთოდები დამადასტურებელი მეთოდები საშუალებას იძლევა ნიმუშში არსებული PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ერთმნიშვნელოვნად (არაორაზროვნად) იდენდიფიკაციისას და რაოდენობრივად განსაზღვრისას, და უზრუნველყოფს სრულ ინფორმაციას კონგენერის დონეზე. ამიტომ ეს მეთოდები საშუალებას იძლევა მაქსიმალური შემცველობისა (ზღვარისა) და მოქმედების ზღუბლების კონტროლისას, სკრინინგის მეთოდებით მიღებული შედეგების დადასტურების ჩათვლით. გარდა ამისა, შედეგები შეიძლება გამოყენებულ იქნეს სხვა მიზნებისთვის, როგორიცაა ცხოველის საკვების მონიტორინგის დაბალი ფონის შემცველობის (ზღვრების) განსაზღვრა, დროის ტენდენციების დაცვა, ექსპოზიციის შეფასება და მონაცემთა ბაზის შექმნა სამოქმედო ზღუბლებისა და მაქსიმალური შემცველობის (ზღვარის) შესაძლო ხელახალი შეფასების მიზნით. ისინი, ასევე მნიშვნელოვანია კონგენერის შაბლონების დასადგენად, შესაძლო დაბინძურების წყაროს დასადგენად. ასეთი მეთოდები იყენებს GC-HRMS-ს. მაქსიმალური შემცველობის (ზღვარის) შესაბამისობის ან შეუსაბამობის დასადასტურებლად, ასევე შეიძლება გამოყენებულ იქნეს GC-MS/MS. 2. ფონი TEQ-ის კონცენტრაციების გამოსათვლელად, მოცემულ ნიმუშში ცალკეული ნივთიერებების კონცენტრაციები გამრავლებულია მათი ტოქსიკური ეკვივალენტობის ფაქტორზე (TEF) (იხ. ამ თავის I ქვეთავის სქოლიო 10) და შემდგომ ჯამდება, რომ დიოქსინის მსგავსი ნაერთების საერთო კონცენტრაცია მივიღოთ, გამოხატული, როგორც TEQ-ები. ამ თავის მიზნებისათვის, ინდივიდუალური კონგენერის რაოდენობრივი შეფასების მიღებული სპეციფიკური ლიმიტი ნიშნავს ანალიტის ყველაზე დაბალ შემცველობას, რომლის გაზომვა შესაძლებელია გონივრული სტატისტიკური სიზუსტით, საიდენტიფიკაციო კრიტერიუმების შესრულებით, როგორც ეს აღწერილია საერთაშორისოდ აღიარებულ სტანდარტებში, მაგალითად, სტანდარტში EN 16215: 2012 (ცხოველის საკვები – დიოქსინებისა და დიოქსინის მსგავსი PCB-ების განსაზღვრა GC-HRMS-ით და ინდიკატორი PCB-ები GC-HRMS-ით) და/ან EPA მეთოდებში 1613 და 1668 შესწორებულ რედაქციაში. ინდივიდუალური კონგენერის რაოდენობრივი შეფასების ლიმიტი შეიძლება განისაზღვროს, როგორც: ა) ანალიტის კონცენტრაცია ნიმუშის ექსტრაქტში, რომელიც აწარმოებს ინსტრუმენტულ რეაქციას ორ განსხვავებულ იონზე, რომლის მონიტორინგი უნდა მოხდეს S/N (სიგნალი/ხმაური) თანაფარდობით 3:1 ნაკლებად ინტენსიური ნედლ მონაცემთა სიგნალისთვის; ბ) ან თუ ტექნიკური მიზეზების გამო სიგნალიდან-ხმაურამდე გაანგარიშება არ იძლევა საიმედო შედეგებს, ყველაზე დაბალი კონცენტრაციის წერტილი საკალიბრო მრუდზე, რომელიც იძლევა მისაღებ (≤ 30%) და თავსებად (იზომება სულ მცირე ნიმუშების ანალიტიკური სერიის დასაწყისში და ბოლოს) გადახრას (დეავიაციას) საშუალო ფარდობითი რეაგირების კოეფიციენტზე, რომელიც გამოითვლება თითოეული სერიის საკალიბრო მრუდის ყველა წერტილზე. რაოდენობრივი შეფასების ლიმიტი (LOQ) გამოითვლება ყველაზე დაბალი კონცენტრაციის წერტილიდან, შიდა სტანდარტების აღდგენისა და ნიმუშის მიღების გათვალისწინებით. ბიოანალიტიკური სკრინინგის მეთოდები არ მოგვცემს შედეგს კონგენერის დონეზე, არამედ მხოლოდ დაგვანახებს(17) TEQ დონეს გამოხატულს BEQ-ში, იმ ფაქტის დასადასტურებლად, რომ არა ყველა ნაერთს, რომელიც არსებობს ნიმუშის ექსტრაქტში, რომლებიც იწვევენ რეაგირებას ტესტში, შეუძლია შეასრულოს ან დააკმაყოფილოს TEQ-პრინციპის ყველა მოთხოვნა. სკრინინგისა და დამადასტურებელი მეთოდების გამოყენება შესაძლებელია მხოლოდ კონკრეტული მატრიცის გასაკონტროლებლად, თუ მეთოდები საკმარისად მგრძნობიარეა შემცველობის (ზღვრების) საიმედო განსაზღვრისთვის მოქმედების ზღურბლზე ან მაქსიმალურ შემცველობაზე. 3. ხარისხის უზრუნველყოფის მოთხოვნები 3.1. ჯვარედინი დაბინძურების თავიდან აცილებისთვის ნიმუშის აღებისა და გამოკვლევის პროცედურის თითოეულ ეტაპზე უნდა იქნეს მიღებული ზომები. 3.2. ნიმუშების შენახვა და ტრანსპორტირება უნდა მოხდეს მინის, ალუმინის, პოლიპროპილენის ან პოლიეთილენის კონტეინერში, რომლებიც შენახვისთვისაა შესაფერისი და არ ახდენს PCDD/F და დიოქსინის მსგავსი PCB-ის შემცველობაზე რაიმე სახის გავლენას. ნიმუშის კონტეინერიდან უნდა მოცილდეს ქაღალდის მტვრის კვალი. 3.3. ნიმუშის შენახვა და ტრანსპორტირება უნდა განხორციელდეს ისე, რომ შენარჩუნდეს ცხოველის საკვების ნიმუშის მთლიანობა (დაურღვევლობა). 3.4. რამდენადაც ეს აუცილებელია, თითოეული ლაბორატორიული ნიმუში წვრილად უნდა დაიფქვას და საფუძვლიანად შეერიოს, იმ პროცედურის მიხედვით, რომელიც ნაჩვენებია სრული ჰომოგენიზაციის მისაღწევად (მაგალითად, ისე დაიფქვას, რომ შესაძლებელი იყოს გაცრა 1 მმ-იან საცერში). ნიმუშები უნდა გაშრეს დაფქვამდე, თუ ტენიანობა ძალიან მაღალია. 3.5. უნდა ჩატარდეს რეაგენტების, მინის ჭურჭლისა და აღჭურვილობის კონტროლი TEQ – ან BEQ-ზე დაფუძნებულ შედეგებზე შესაძლო გავლენისთვის. 3.6. საკონტროლო გამოკვლევა უნდა ჩატარდეს მთლიანი გამოკვლევის პროცედურის ჩატარებით, მხოლოდ ნიმუშის გამორიცხვით. 3.7. ბიოანალიტიკური მეთოდებისთვის, ყველა მინის ჭურჭელი და გამხსნელი, რომლებიც გამოიყენება გამოკვლევაში, უნდა შემოწმდეს ნაერთების არარსებობაზე, რომლებიც ხელს უშლიან სამიზნე ნაერთების გამოვლენას სამუშაო დიაპაზონში. მინის ჭურჭელი უნდა გაირეცხოს გამხსნელებით ან გათბეს შესაფერისი PCDD/F-ების ტემპერატურაზე, დიოქსინის მსგავსი ნაერთებისა და მისი ზედაპირის შემაფერხებელი ნაერთების კვალის მოსაცილებლად. 3.8. ნიმუშების ის რაოდენობა, რომელიც გამოიყენება ექსტრაჰირებისათვის, საკმარისია მოთხოვნების შესასრულებლად დაბალი სამუშაო დიაპაზონთან მიმართებაში, მაქსიმალური შემცველობის (ზღვარის) კონცენტრაციების ან მოქმედების ზღურბლის ჩათვლით. 3.9. კონკრეტული ნიმუშის მოსამზადებელი პროცედურები, რომლებიც გამოიყენება განხილვას დაქვემდებარებული პროდუქტებისთვის, უნდა შეესაბამებოდეს საერთაშორისო დონეზე მიღებულ სახელმძღვანელო მითითებებს.
4. მოთხოვნები ლაბორატორიისთვის 4.1. ლაბორატორიები აკრედიტებული უნდა იყოს აღიარებული ორგანოს მიერ, რომელიც მუშაობს ISO 58-ე სახელმძღვანელოს შესაბამისად, იმის უზრუნველსაყოფად, რომ ისინი იყენებენ გამოკვლევის ხარისხის უზრუნველყოფას. ლაბორატორიები უნდა იყოს აკრედიტირებული EN ISO/IEC 17025 სტანდარტის შესაბამისად. განუსაზღვრელობის განსაზღვრის შეფასების ტექნიკურ სახელმძღვანელოში აღწერილი პრინციპები და PCDD/F და PCB გამოკვლევის რაოდენობრივი შეფასების ზღვრები უნდა იყოს დაცული, როდესაც ეს აუცილებელია.18 4.2. ლაბორატორიული გამოცდილება უნდა დადასტურდეს უწყვეტი წარმატებული მონაწილეობით ლაბორატორიათშორის გამოკვლევებში PCDD/F და დიოქსინის მსგავსი PCB-ების დასადგენად შესაბამის ცხოველის საკვებ მატრიცებში და კონცენტრაციის დიაპაზონში. 4.3. ლაბორატორიები, რომლებიც იყენებენ სკრინინგის მეთოდებს ნიმუშების რუტინული კონტროლისთვის, უნდა დაამყარონ მჭიდრო თანამშრომლობა იმ ლაბორატორიებთან, რომლებიც იყენებენ დამადასტურებელ მეთოდს, როგორც ხარისხის კონტროლისთვის, ისე საეჭვო ნიმუშების გამოკვლევის შედეგის დასადასტურებლად.
5. ძირითადი მოთხოვნები, რომლებიც უნდა დაკმაყოფილდეს დიოქსინების (PCDD/Fs) და დიოქსინის მსგავსი PCB-ის გამოკვლევის პროცედურით 5.1. დაბალი სამუშაო დიაპაზონი და რაოდენობრივი შეფასების ზღვრები PCDD/F-ებისთვის, გამოსავლენი რაოდენობა უნდა იყოს ზედა ფემტოგრამის (10-15 გრ) დიაპაზონში, ამ ზოგიერთი ნაერთების უკიდურესი ტოქსიკურობის გამო. PCB კონგენერთა უმეტესობისთვის ნანოგრამის (10–9 გრ) დიაპაზონში რაოდენობრივი ზღვარი უკვე საკმარისია. უფრო ტოქსიკური დიოქსინის მსგავსი PCB კონგენერების გაზომვისთვის (კერძოდ, არაორთოჩანაცვლებითი კონგენერებისათვის), სამუშაო დიაპაზონის ქვედა ბოლომ უნდა მიაღწიოს პიკოგრამის დაბალ ზღვარს (10-12 გ). ყველა სხვა PCB კონგენერებისათვის საკმარისია ნანოგრამის (10–9 გრ) დიაპაზონში რაოდენობრივი განსაზღვრის ზღვარი. 5.2. მაღალი შერჩევითობა (სპეციფიკურობა) 5.2.1. საჭიროა განსხვავება PCDD/F და დიოქსინის მსგავსი PCB-ებს შორის და სხვა, კოექსტრაქციულ და, შესაძლოა, ხელშემშლელ ნაერთებს შორის, რომლებიც კონცენტრაციებში რამდენიმე სიდიდით მაღლაა, ვიდრე გამოსაკვლევი ანალიტები. GC-MS მეთოდებისათვის საჭიროა დიფერენციაცია სხვადასხვა კონგენერებს შორის, მაგალითად, ტოქსიკურ (მაგალითად, ჩვიდმეტი 2,3,7,8-ჩანაცვლებული PCDD/F-ები, და თორმეტი დიოქსინის მსგავსი PCB-ები) და სხვა კონგენერებს შორის. 5.2.2. ბიოანალიტიკურ მეთოდებს უნდა შეეძლოს სამიზნე ნაერთების გამოვლენა, როგორც PCDD/F-ებისა და/ან დიოქსინის მსგავსი PCB-ების ჯამი. ნიმუშის გაწმენდა მიზნად ისახავს ცრუ შეუსაბამო შედეგების გამომწვევი ნაერთების ან ნაერთების მოშორებას, რომლებმაც შეიძლება შეამცირონ რეაგირება, გამოიწვიოს ცრუ შესაბამისობის შედეგები. 5.3. მაღალი აკურატულობა (ნამდვილობა და სიზუსტე, ბიოანალიზის აშკარა აღდგენა) 5.3.1. GC-MS-ის მეთოდებისთვის განსაზღვრებამ უნდა უზრუნველყოს ნიმუშის ნამდვილი კონცენტრაციის სწორი შეფასება. საჭიროა მაღალი სიზუსტე, რათა თავიდან იქნეს აცილებული ნიმუშის გამოკვლევის შედეგების უარყოფა, განსაზღვრული TEQ დონის დაბალი სანდოობის საფუძველზე. აკურატულობა გამოიხატება, როგორც ნამდვილობა (სხვაობა სერტიფიცირებულ მასალაში ანალიტისთვის გაზომილ საშუალო მნიშვნელობასა და მის სერტიფიცირებულ მნიშვნელობას (რომელიც გამოხატულია, როგორც მნიშვნელობის პროცენტი) შორის და სიზუსტე (RSDR ფარდობითი სტანდარტული გადახრა გამოითვლება განმეორებადობის პირობებში წარმოქმნილი შედეგებიდან). 5.3.2. ბიოანალიტიკური მეთოდებისათვის განისაზღვრება ბიოანალიზის აშკარა აღდგენა. ბიოანალიზის აშკარა აღდგენა ნიშნავს BEQ დონეს, რომელიც გამოითვლება TCDD ან PCB 126 საკალიბრო მრუდიდან, რომელიც შესწორებულია სუფთა ნაწილისთვის და შემდეგ იყოფა TEQ-ის დონით, რომელიც განისაზღვრება დამადასტურებელი მეთოდით. ის მიზნად ისახავს ისეთი ფაქტორების გამოსწორებას, როგორიცაა PCDD/F და დიოქსინის მსგავსი ნაერთების დაკარგვა მოპოვებისა და გაწმენდის ეტაპებზე, თანაექსტრაჰირებული ნაერთები, რომლებიც ზრდიან ან ამცირებენ რეაგირებას (აგონისტური და ანტაგონისტური ეფექტები), მრუდის ხარისხსს ან განსხვავებებს TEF და ფარდობით პოტენციის მნიშვნელობებს (REP) შორის. ბიოანალიზის აშკარა აღდგენა გამოითვლება შესაფერისი ეტალონური ნიმუშებიდან, წარმომადგენლობითი კონგენერული ნიმუშებით ინტერესის ზღვარის ფარგლებში. 5.4. ვალიდაცია მაქსიმალური შემცველობის (ზღვარის) დიაპაზონში და ხარისხის კონტროლის ზოგადი ზომები 5.4.1. ლაბორატორიებმა უნდა წარმოაჩინონ მეთოდის შესრულება მაქსიმალური შემცველობის (ზღვარის) დიაპაზონში, მაგალითად, 0,5x, 1x და 2x მაქსიმალურ შემცველობაზე, განმეორებითი გამოკვლევისთვის, მისაღები ვარიაციის კოეფიციენტით, ვალიდაციის პროცედურის და რუტინული გამოკვლევის დროს. 5.4.2. რეგულარული სუფთა კონტროლი და დანამატებიანი ექსპერიმენტები ან სუფთა ნიმუშების გამოკვლევა (სასურველია, თუ არსებობს, დამოწმებული ეტალონური მასალა) უნდა შესრულდეს, როგორც ხარისხის შიდა კონტროლის ღონისძიებები. უნდა მოხდეს სუფთა კონტროლის, დანამატებიანი ექსპერიმენტის ან სუფთა ნიმუშების გამოკვლევის ხარისხის კონტროლის ცხრილების ანგარიშში ჩაწერა და შემოწმება, რათა დავრწმუნდეთ, რომ გამოკვლევა შეესაბამება მოთხოვნებს. 5.5. რაოდენობრივი განსაზღვრის ლიმიტები (ზღვრები) 5.5.1. ბიოანალიტიკური სკრინინგის მეთოდისთვის რაოდენობრივი განსაზღვრის ლიმიტის (ზღვარის) დადგენა არ არის შეუცვლელი მოთხოვნა, მაგრამ ამ მეთოდით უნდა დამტკიცდეს, რომ მას შეუძლია განასხვაოს სუფთა და ზღვრული მნიშვნელობა (cut-off value). BEQ დონის უზრუნველყოფისას უნდა დადგინდეს საანგარიშო დონე ნიმუშებისთვის, რომლებიც აჩვენებენ ამ დონეზე დაბალ რეაგირებას. საანგარიშო დონე უნდა იყოს ნაჩვენები, რათა პროცედურის სუფთა ნიმუშებისგან იყოს განსხვავებული, სულ მცირე სამი ფაქტორით, სამუშაო დიაპაზონზე დაბალი რეაქციით. ამიტომ იგი უნდა გამოითვალოს სამიზნე ნაერთების შემცველი ნიმუშებიდან, რომელიც იმყოფება საჭირო მინიმალური დონის ფარგლებში, და არა S/N რაციოდან ან სუფთა ანალიზიდან. 5.5.2. დამადასტურებელი მეთოდის LOQ უნდა იყოს მაქსიმალური შემცველობის მეხუთედი. 5.6. ანალიტიკური კრიტერიუმები დამადასტურებელი ან სკრინინგის მეთოდებიდან, საიმედო შედეგების მისაღწევად, შემდეგი კრიტერიუმები უნდა დაკმაყოფილდეს TEQ ან BEQ მნიშვნელობის მაქსიმალური შემცველობის დიაპაზონში, მიუხედავად იმისა განისაზღვრება იგი, როგორც მთლიანი TEQ ან მთლიანი BEQ (როგორც PCDD/Fs და დიოქსინი – PCB– ების მსგავსად) ან ცალკე PCDD/F და დიოქსინის მსგავსი PCB:
5.7. სკრინინგის მეთოდების სპეციფიკური მოთხოვნები 5.7.1. სკრინინგისთვის შეიძლება გამოყენებულ იქნეს როგორც GC-MS, ასევე ბიოანალიტიკური მეთოდები. GC-MS მეთოდებისთვის უნდა დაკმაყოფილდეს ამ ქვეთავის მე-6 პუნქტით გათვალისწინებული მოთხოვნები. უჯრედზე დაფუძნებული ბიოანალიტიკური მეთოდებისათვის სპეციფიკური მოთხოვნები მოცემულია ამ ქვეთავის მე-7 პუნქტში. 5.7.2. ლაბორატორიებმა, რომლებიც იყენებენ სკრინინგის მეთოდებს ნიმუშების რუტინული კონტროლისთვის, მჭიდრო თანამშრომლობა უნდა დაამყარონ იმ ლაბორატორიებთან, რომლებიც იყენებენ დამადასტურებელ მეთოდებს. 5.7.3. სკრინინგის მეთოდის მუშაობის ვერიფიკაცია საჭიროა რუტინული გამოკვლევის დროს, ხარისხის ანალიტიკური კონტროლისა და მიმდინარე მეთოდის ვალიდაციით. უნდა არსებობდეს უწყვეტი პროგრამა შესაბამისობის მქონე ნიმუშების შედეგების კონტროლისთვის. 5.7.4. შეამოწმეთ უჯრედების რეაქციისა და ციტოტოქსიურობის შესაძლო ჩახშობა: ნიმუშის ექსტრაქტების 20% უნდა გაიზომოს რუტინულ სკრინინგში 2,3,7,8-TCDD-ის ან მის გარეშე დამატებით, მაქსიმალური შემცველობის ან მოქმედების ბარიერის შესაბამისად, იმის შესამოწმებლად, შესაძლებელია თუ არა რეაქციის აღკვეთა ნიმუშის ექსტრაქტში არსებული ხელშემშლელი ნივთიერებებით. დანამატებიანი ნიმუშის (spiked sample) გაზომილ კონცენტრაციას უნდა შევადაროთ დაუმატებელი ექსტრაქტის (unspiked extract) კონცენტრაციის ჯამს პლიუს დანამატებიანი კონცენტრაცია. თუ გაზომილი კონცენტრაცია 25%-ზე მეტია ვიდრე დაანგარიშებული (ჯამური) კონცენტრაცია, ეს არის ჩახშობის მაჩვენებელი, პოტენციური სიგნალი, და შესაბამისი ნიმუში იგზავნება GC-HRMS დამადასტურებელი გამოკვლევის მიზნით. ხარისხის კონტროლის ცხრილებში უნდა მოხდეს შედეგების მონიტორინგი. 5.7.5. შესაბამისობის შედეგის მქონე ნიმუშების ხარისხის კონტროლი: შესაბამისობის შედეგის მქონე ნიმუშების დაახლოებით 2-დან 10% -მდე, უნდა დადასტურდეს GC/HRMS-ით, ნიმუშის მატრიცისა და ლაბორატორიულ გამოცდილებიდან გამომდინარე. 5.7.6. ხარისხის კონტროლის მონაცემების მიხედვით ცრუ-შესაბამისი მაჩვენებლების განსაზღვრა: უნდა განისაზღვროს ცრუ-შესაბამისი შედეგების მაჩვენებელი სკრინინგის ნიმუშის ქვედა და ზედა მაქსიმუმის ზღვარიდან ან მოქმედების ზღურბლიდან. რეალური ცრუ-შესაბამისი მაჩვენებლები არ უნდა აღემატებოდეს 5%-ს. როდესაც შესაბამისი მინიმალური ნიმუშების ხარისხის კონტროლიდან შესაძლებელია არანაკლებ 20 დადასტურებული შედეგი, დასკვნები ცრუ-შესაბამისი მაჩვენებელზე უნდა გაკეთდეს ამ მონაცემთა ბაზიდან. რგოლურ გამოცდაში ან დაბინძურების ინციდენტის დროს გამოკვლეული ნიმუშების შედეგები, რომლებიც მოიცავს კონცენტრაციის დიაპაზონს, მაგალითად, მაქსიმუმ 2-ჯერ მაქსიმალურ ზღვარს (ML), ასევე შეიძლება მოიცავდეს სულ მცირე 20 შედეგს, ცრუ-შესაბამისი მაჩვენებლის შესაფასებლად. ნიმუშები მოიცავს ყველაზე ხშირად კონგენერულ ნიმუშებს, რომლებიც წარმოადგენენ სხვადასხვა წყაროებს. მიუხედავად იმისა, რომ სკრინინგის ანალიზი უპირატესად მიზნად ისახავს მოქმედების ზღურბლზე მეტი ნიმუშების გამოვლენას, ცრუ-შესაბამისობის მაჩვენებლების განსაზღვრის კრიტერიუმი არის მაქსიმალური ზღვარი, დამადასტურებელი მეთოდის განუსაზღვრელობის გაფართოებული განსაზღვრის გათვალისწინებით. 5.7.7. სკრინინგის შედეგად მიღებული პოტენციური შეუსაბამო ნიმუშები ყოველთვის უნდა იქნეს ვერიფიცირებული ორიგინალი ნიმუშის ხელახალი გამოკვლევით, დადასტურების მეთოდით. ეს ნიმუშები ასევე შეიძლება გამოყენებულ იქნეს ცრუ-შეუსაბამო შედეგების სიხშირის შესაფასებლად. სკრინინგის მეთოდებისათვის, ცრუ შეუსაბამო შედეგების სიხშირე უნდა იყოს დადასტურებული გამოკვლევის შედეგად დადასტურებული შედეგების წილი, მაშინ როდესაც წინა სკრინინგში ნიმუში გამოცხადებული იქნა პოტენციურად შეუსაბამოდ. სკრინინგის მეთოდის უპირატესობათა შეფასება უნდა ეფუძნებოდეს ცრუ-შეუსაბამო ნიმუშების შედარებას შემოწმებულ ნიმუშების საერთო რაოდენობასთან. ეს მაჩვენებელი საკმარისად დაბალი უნდა იყოს, რათა სკრინინგის საშუალების გამოყენება იყოს უპირატესი. 5.7.8. ვალიდაციის პირობებში, ბიოანალიტიკური მეთოდებმა უნდა უზრუნველყოს TEQ დონის მართებული მითითება, გამოთვლილი და გამოხატულის BEQ-ით. ასევე განმეორებით პირობებში ჩატარებული ბიოანალიტიკური მეთოდებისთვის, ლაბორატორიატთაშორისი RSDr, როგორც წესი, უფრო მცირე იქნება, ვიდრე რეპროდუქციულობის (შედეგიანობის) პირობებში (RSDR). 6. GC-MS მეთოდებთან დაკავშირებული სპეციალური მოთხოვნები, რომელთა დაცვა აუცილებელია სკრინინგის ან დადასტურების მიზნით 6.1. მისაღები განსხვავებები WHO-TEQ-ის ზედა და ქვედა ზღვარის შედეგებს შორის სხვაობა ზედა და ქვედა ზღვრებს შორის არ უნდა აღემატებოდეს 20%-ს, მაქსიმალური შემცველობის (ზღვარის) გადაჭარბების ან საჭიროების შემთხვევაში, მოქმედების ზღურბლის დასადასტურებლად. 6.2. აღდგენის კონტროლი 6.2.1. 13C ნიშანდებული 2,3,7,8-ქლორის შემცვლელი შიდა PCDD/F სტანდარტებისა და 13C ნიშანდებული შიდა დიოქსინის მსგავსი PCB სტანდარტების დამატება უნდა განხორციელდეს გამოკვლევის მეთოდის დასაწყისში, მაგ. ექსტრაჰირებამდე გამოკვლევის პროცედურის ვალიდაციისათვის. სულ მცირე ერთი კონგენერი უნდა დაემატოს თითოეულ ტეტრადან ოქტა-ქლორიებული ჰომოლოგიური ჯგუფისათვის PCDD/F-ებისთვის და სულ მცირე ერთი კონგენერი თითოეული ჰომოლოგიური ჯგუფისათვის დიოქსინის მსგავსი PCB-ებისთვის (ალტერნატიულად, სულ მცირე ერთი კონგენერის თითოეული მას-სპექტრომეტრიული შერჩეული იონების ჩამწერი ფუნქციისთვის, რომელიც გამოიყენება PCDD/F და დიოქსინის მსგავსი PCB-ების მონიტორინგისთვის). დამადასტურებელი მეთოდების შემთხვევაში, გამოყენებული უნდა იყოს ყველა 17 13C ნიშანდებული 2,3,7,8-ჩანაცვლებული შიდა PCDD/F სტანდარტები და ყველა 12 13C ნიშანდებული შიდა დიოქსინის მსგავსი PCB სტანდარტი. 6.2.2. ფარდობითი რეაგირების ფაქტორები ასევე უნდა განისაზღვროს იმ კონგერებისთვის, რომელთათვისაც არ ემატება 13C ნიშანდებული ანალოგი შესაბამისი საკალიბრო ხსნარების გამოყენებით. 6.2.3. მცენარეული და ცხოველური წარმოშობის ცხოველის საკვებისთვის, რომელიც შეიცავს 10%-ზე ნაკლებ ცხიმს, ექსტრაჰირებამდე სავალდებულოა შიდა სტანდარტების დამატება. ცხოველური წარმოშობის ცხოველის საკვებისთვის, რომელიც შეიცავს 10%-ზე მეტ ცხიმს, შიდა სტანდარტები დაემატება ცხიმის ექსტრაჰირებამდე ან მის შემდეგ. ექსტრაჰირების ეფექტურობის შესაბამისი შემოწმება (ვალიდაცია) უნდა განხორციელდეს იმის მიხედვით, თუ რა ეტაპზეა წარმოდგენილი შიდა სტანდარტები. 6.2.4. GC-MS ანალიზამდე უნდა დაემატოს 1 ან 2 აღდგენის (სუროგატი) სტანდარტ(ებ)ი. 6.2.5. საჭიროა აღდგენის კონტროლი. დამადასტურებელი მეთოდებისთვის ინდივიდუალური შიდა სტანდარტების აღდგენა უნდა იყოს 60-დან 120%-ის ფარგლებში. ცალკეული კონგენერისთვის, განსაკუთრებით ზოგიერთ ჰეპტა-ქლორიანი და ოქტა-ქლორიანი დიბენზო – p – დიოქსინებისა და დიბენზოფურანებისთვის, ქვედა ან მაღალი აღდგენა მიიღება იმ პირობით, რომ TEQ-ის მნიშვნელობაში მათი კონტრიბუცია არ უნდა აღემატებოდეს TEQ-ის მთლიანი მნიშვნელობის 10%-ს (PCDD/F და დიოქსინის მსგავსი PCB-ების ჯამზე დაყრდნობით). GC-MS სკრინინგის მეთოდებისთვის, აღდგენა უნდა იყოს 30-დან 140%-ის ფარგლებში. 6.3. ხელშემშლელი ნივთიერებების მოცილება – PCDD/F-ის განცალკევება ქლორირებული ნაერთებისგან, როგორიცაა არადიოქსინის მსგავსი PCB-ები და ქლორირებული დიფენილის ეთერები, უნდა განხორციელდეს შესაფერისი ქრომატოგრაფიული ტექნიკით (სასურველია ფლორისილით, ალუმინის ან/და ნახშირბადის სვეტით). – იზომერების გაზ-ქრომატოგრაფიული გამიჯვნა უნდა იყოს <25% პიკიდან პიკამდე, 1,2,3,4,7,8-HxCDF და 1,2,3,6,7,8-HxCDF შორის. 6.4. სტანდარტული მრუდით დაკალიბრება საკალიბრო მრუდის დიაპაზონი მოიცავს მაქსიმალური ზღვარის ან მოქმედების ზღურბლის შესაბამის დიაპაზონს. 6.5. დამადასტურებელი მეთოდების სპეციფიური კრიტერიუმები – GC-HRMS-ისთვის: HRMS-ში, გარჩევადობა, როგორც წესი, მთლიანი მასური დიაპაზონისთვის 10% ველზე, უნდა იყოს 10 000-ზე მეტი ან თანაბარი. შემდგომი იდენტიფიკაციისა და დადასტურების კრიტერიუმების შესრულება, როგორც ეს აღწერილია საერთაშორისოდ აღიარებულ სტანდარტებში, მაგალითად, სტანდარტში EN 16215: 2012 (ცხოველის საკვები – დიოქსინებისა და დიოქსინის მსგავსი PCB-ების განსაზღვრა GC-HRMS-ით და ინდიკატორი PCB-ების მიერ GC-HRMS– ით) და/ან EPA მეთოდებში 1613 და 1668 შესწორებებით. – GC-MS/MS-სთვის: სულმ მცირე 2 სპეციფიკური პრეკურსორ-იონის მონიტორინგი, რომელთაგან თითოეულს აქვს ერთი სპეციფიკური შესაბამისი გარდამავალი პროდუქტის იონი, ყველა ეტიკეტირებული და არაეტიკეტირებული ანალიტისთვის გამოკვლევის ფარგლებში. ± 15% იონის ფარდობითი ინტენსივობის მაქსიმალური დასაშვები ტოლერანტობა შერჩეული გარდამავალი პროდუქტის იონებისთვის, გამოანგარიშებულ ან გაზომილ მნიშვნელობებთან შედარებით (საშუალო საკალიბრო სტანდარტებიდან), გამოიყენება იდენტური MS/MS პირობები, განსაკუთრებით შეჯახების ენერგიისა და გაზების შეჯახების წნევა, ანალიტის თითოეული გადასვლისთვის. თითოეული კვადრუპოლის გარჩევადობის უნარი უნდა იყოს თანაბარი ან უკეთესი, ვიდრე ზოგაგად ხელსაწყოსი (მასის გარჩევადობის უნარი: საკმარისი გარჩევადობა, რომ ერთი მასის ერთეულის დაშორებით მყოფო ორი პიკის მწვერვალები ცალ-ცალკე გამოიყოს), რათა შევამციროთ პოტენციური ხელის შემშლელი პირობები გამოსაკვლევ ანალიტებზე. შემდგომი კრიტერიუმების შესრულება, როგორც ეს აღწერილია საერთაშორისოდ აღიარებულ სტანდარტებში, მაგალითად, სტანდარტში EN 16215: 2012 (ცხოველის საკვები – დიოქსინებისა და დიოქსინის მსგავსი PCB-ების განსაზღვრა GC-HRMS-ით და ინდიკატორი PCB-ების მიერ GC-HRMS– ით) და/ან EPA მეთოდებში 1613 და 1668 შესწორებებით, GC-HRMS-ის გამოყენების ვალდებულების გარდა. 7. სპეციფიკური მოთხოვნები ბიოანალიტიკური მეთოდების მიმართ ბიოანალიტიკური მეთოდები არის მეთოდები, რომლებიც დაფუძნებულია ბიოლოგიური პრინციპების გამოყენებაზე, როგორიცაა უჯრედოვანი, რეცეპტორების ან იმუნო გამოკვლევები. ეს პუნქტი ზოგადად ადგენს მოთხოვნებს ბიოანალიტიკური მეთოდების მიმართ. სკრინინგის მეთოდი, პრინციპში, ახდენს ნიმუშის კლასიფიკაციას, როგორც შესაბამისს ან სავარაუდოდ შეუსაბამოს (საეჭვოს). ამისათვის, გამოთვლილი BEQ დონე დარდება ზღვრულ (cut-off value) მნიშვნელობასთან (იხ. ამ ქვეთავის პუნქტი 7.3). ზღვრული მნიშვნელობის (cut-off value) ქვემოთ გამოვლენილი ნიმუშები ცხადდება შესაბამისად, ხოლო ის ნიმუშები, რომლებიც თანაბარია ან მეტია ზღვრულ მნიშვნელობაზე (cut-off value), ესაჭიროება გამოკვლევა დამადასტურებელი მეთოდით. პრაქტიკაში, BEQ დონე, რომელიც მაქსიმალური შემცველობის მესამედს შეესაბამება, შეიძლება გახდეს ზღვრული (cut-off value) მნიშვნელობა, იმ პირობით, თუკი უზრუნველყოფილი იქნება 5%-ზე ნაკლები ცრუ შესაბამისობის მაჩვენებელი და ცრუ შეუსაბამო შედეგების მისაღები მაჩვენებელი. ცალკეული მაქსიმალური შემცველობის მქონე PCDD/F-ებისთვის, PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამისთვის, ნიმუშების შესაბამისობის შემოწმება ფრაქციის გარეშე მოითხოვს PCDD/F-ების შესაბამის ბიოანალიზის ზღვრულ (cut-off value) მნიშვნელობებს. იმ ნიმუშების შემოწმების მიზნით, რომლებიც აღემატებიან მოქმედების ზღრუბლს, შესაბამისი მოქმედების ზღურბლის შესაბამისი პროცენტი უნდა იქნეს გამოყენებული, როგორც ზღვრული (cut-off value) მნიშვნელობა. თუ მითითებული დონე გამოხატულია BEQ-ებში, ნიმუშის შედეგები უნდა იყოს სამუშაო დიაპაზონში და უნდა აღემატებოდეს საანგარიშო ზღვარს (იხ. ამ ქვეთავის პუნქტი 7.1.1 და 7.1.6). 7.1. ტესტის პასუხის შეფასება 7.1.1. ზოგადი მოთხოვნები – TCDD საკალიბრო მრუდიდან, კონცენტრაციების გაანგარიშებისას, მრუდის მაღალი ბოლოს მნიშვნელობები აჩვენებს მაღალ ვარიაციას (ვარიაციის მაღალი კოეფიციენტი (CV)). სამუშაო დიაპაზონი არის ის ტერიტორია, სადაც ეს CV ნაკლებია 15%-ზე. სამუშაო დიაპაზონის ქვედა ზღვარი (საანგარიშო ზღვარი) უნდა განისაზღვროს საკონტროლო (სუფთა) პროცედურაზე სულ მცირე სამი კოეფიციენტით მეტით. სამუშაო დიაპაზონის ზედა ბოლო ჩვეულებრივ წარმოდგენილია EC70 მნიშვნელობით (მაქსიმალური ეფექტური კონცენტრაციის 70%), მაგრამ უფრო დაბალია, თუ CV ამ დიაპაზონში 15%-ზე მაღალია. სამუშაო დიაპაზონი უნდა დადგინდეს ვალიდაციის დროს. ზღვრული (cut-off value) მნიშვნელობები (იხ. ამ ქვეთავის პუნქტი 7.3) კარგად უნდა იყოს მოქცეული სამუშაო დიაპაზონში. – სტანდარტული ხსნარები და ექსტრაქტების ნიმუშები უნდა შემოწმდეს სამ ეგზემპლარში ან არანაკლებ ორ ეგზემპლარში (დუბლიკატში). დუბლიკატების გამოყენებისას, სტანდარტული ხსნარი ან საკონტროლო ექსტრაქტი, რომელიც ტესტირებულია ფირფიტაზე დაყოფილ ოთხ-ექვს ფოსოში, უნდა წარმოქმნას რეაქცია ან კონცენტრაცია (შესაძლებელია მხოლოდ სამუშაო დიაპაზონში) CV <15%-ზე დაყრდნობით. 7.1.2. დაკალიბრება 7.1.2.1. დაკალიბრება სტანდარტული მრუდით – ნიმუშებში შემცველობის (ზღვარის) შეფასება უნდა მოხდეს, TCDD (ან PCB 126 ან PCDD/PCDF/დიოქსინის მსგავსი PCB სტანდარტული ნარევის) ტესტის პასუხის საკალიბრო მრუდთან შედარების გზით, რათა გამოვთვალოთ BEQ დონე ექსტრაქტში და შემდეგ ნიმუშში. – საკალიბრო მრუდები უნდა შეიცავდეს 8 – 12 კონცენტრაციას (მინიმუმ დუბლიკატებში), საკმარისი კონცენტრაციებით მრუდის ქვედა ნაწილში (სამუშაო დიაპაზონში). განსაკუთრებული ყურადღება უნდა დაეთმოს მრუდის მორგების (curve-fit) ხარისხს ოპერაციულ დიაპაზონში. ამრიგად, R2 მნიშვნელობას აქვს მცირე ან საერთოდ არ აქვს მნიშვნელობა, არაწრფივი რეგრესიის ვარგისიანობის ხარისხის შეფასებისას. საუკეთესო ვარგისიანობა მიიღწევა მრუდის საოპერაციო დიაპაზონში გამოანგარიშებულ და დაკვირვებულ დონეთა სხვაობის შემცირებით, მაგალითად, ნაშთის (ნარჩენების) კვადრატების ჯამის მინიმიზაციით (უმცირეს კვადრატთა მეთოდი). – ნიმუშის ექსტრაქტში შეფასებული შემცველობა (ზღვარი) შემდგომში უნდა შესწორდეს BEQ შემცველობისთვის (ზღვარისთვის), რომელიც გამოითვლება მატრიცის ან გამხსნელის საკონტროლო (სუფთა) ნიმუშისთვის (გამოყენებული გამხსნელებისა და ქიმიკატების მინარევების გასაცნობად) და აშკარა აღდგენისთვის (გამოითვლება შესაფერისი ეტალონური ნიმუშების BEQ შემცველობიდან (ზღვარიდან), წარმომადგენლობითი კონგენერის ნიმუშებით მაქსიმალური შემცველობის ან მოქმედების ზღვარის ირგვლივ). აღდგენის შესწორების შესასრულებლად, აშკარა აღდგენა უნდა მოხდეს საჭირო დიაპაზონში (იხ. ამ ქვეთავის პუნქტი 7.1.4). აღდგენის კორექციისთვის გამოყენებული ეტალონური ნიმუშები უნდა შეესაბამებოდეს ამ ქვეთავის 7.2-ე პუნქტით დადგენილ მოთხოვნებს. 7.1.2.2. დაკალიბრება ეტალონური ნიმუშებით გარდა ამისა, შეიძლება გამოყენებულ იქნეს საკალიბრო მრუდი სულ მცირე ოთხი ეტალონური ნიმუშისგან (იხ. ამ ქვეთავის პუნქტი 7.2.4): ერთი მატრიცის საკონტროლო (სუფთა), პლუს სამი ეტალონური ნიმუში 0,5x, 1x და 2x მაქსიმალური შემცველობით ან მოქმედების ზღრუბლით შეიძლება იქნეს გამოყენებული, რომელიც გამორიცხავს კორექტირების საჭიროებას საკონტროლო ნიმუშოსთვის (სუფთასთვის) და აღდგენას, თუ ეტალონური ნიმუშების მატრიცული თვისებები ემთხვევა უცნობ ნიმუშებს. ამ შემთხვევაში, ტესტის პასუხი, რომელიც შეესაბამება მაქსიმალური შემცველობის ორ მესამედს (იხ. ამ ქვეთავის პუნქტი 7.3), შეიძლება გამოითვალოს უშუალოდ ამ ნიმუშებიდან და გამოყენებულ იქნეს, როგორც ზღვრული (cut-off value) მნიშვნელობა. იმ ნიმუშების შემოწმების მიზნით, რომლებიც აღემატებიან მოქმედების ზღრუბლს, შესაბამისი მოქმედების ზღურბლის შესაბამისი პროცენტი, უნდა იქნეს გამოყენებული როგორც ზღვრული (cut-off value) მნიშვნელობა. 7.1.3. PCDD/F და დიოქსინის მსგავსი PCB-ების ცალკეული განსაზღვრა ექსტრაქტები შეიძლება დაიყოს ფრაქციებად, რომლებიც შეიცავს PCDD/F და დიოქსინის მსგავს PCB-ს, რაც საშუალებას იძლევა ცალკე აღნიშნოს PCDD/F-ისა და დიოქსინის მსგავსი PCB TEQ შემცველობები (ზღვრები) (BEQ-ში). PCB 126 სტანდარტული საკალიბრო მრუდი უპირატესად უნდა იქნეს გამოყენებული დიოქსინის მსგავსი PCB-ის შემცველი ფრაქციის შედეგების შესაფასებლად. 7.1.4. ბიოანალიზის აშკარა აღდგენა „ბიოანალიზის აშკარა აღდგენა“ უნდა გამოითვალოს შესაფერისი ეტალონური ნიმუშებიდან, წარმოდგენილ კონგენერის ნიმუშებით მაქსიმალური შემცველობის ან მოქმედების ზღურბლის ფარგლებში და რომელიც გამოხატულია BEQ შემცველობის (ზღვარის) პროცენტული მაჩვენებელით TEQ შემცველობასთან (ზღვართან) შედარებით. გამოყენებული ანალიზისა და TEF-ების19 ტიპებიდან გამომდინარე, დიოქსინის მსგავსი PCB-ების TEF-სა და REP-ის ფაქტორებს შორის განსხვავებამ შეიძლება გამოიწვიოს დიოქსინის მსგავსი PCB-ების დაბალი „აშკარა აღდგენა“ PCDD/F-ებთან შედარებით. ამიტომ, თუ PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ცალკეული განსაზღვრა განხორციელდა, უნდა მოხდეს ბიოანალიზის აშკარა აღდგენა: დიოქსინის მსგავსი PCB-ებისთვის 20%-დან 60%-მდე, PCDD/F-ებისთვის 50%-დან 130%-მდე (დიაპაზონები ვრცელდება TCDD საკალიბრო მრუდზე). იმის გამო, რომ დიოქსინის მსგავსი PCB-ების კონტრიბუცია PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამში შეიძლება განსხვავდებოდეს სხვადასხვა მატრიცებსა და ნიმუშებს შორის, ბიოანალიზის აშკარა აღდგენა PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამისთვის ასახავს ამ დიაპაზონებს და უნდა იყოს 30%-დან 130%-მდე. არსებითად შესწორებული TEF მნიშვნელობების ნებისმიერი გავლენა კანონმდებლობაში PCDD/F და დიოქსინის მსგავსი PCB-ებზე, საჭიროებს ამ დიაპაზონის გადახედვას. 7.1.5. აღდგენის კონტროლი გაწმენდის მიზნით გაწმენდის დროს ნაერთების დაკარგვა უნდა შემოწმდეს ვალიდაციისას. საკონტროლო ნიმუში სხვადასხვა კონგენერების ნარევების დამატებით უნდა წარედგინოს გაწმენდისთვის (სულ მცირე N = 3) და დადასტურების მეთოდით უნდა შემოწმდეს აღდგენა და ვარიაცია. აღდგენა უნდა იყოს 60%-დან 120%-მდე, განსაკუთრებით იმ კონგენერებისთვის, რომლებსაც 10%-ზე მეტი კონტრიბუცია შეაქვთ TEQ-ის შემცველობაზე (ზღვარზე) სხვადასხვა ნარევებში. 7.1.6. ანგარიშგების ზღვრები BEQ ზღვარის მოხსენებისას, ანგარიშგების ზღვრები უნდა განისაზღვროს შესაბამისი მატრიცული ნიმუშებიდან, რომლებიც მოიცავს ტიპური კონგენერულ ნიმუშებს, მაგრამ არა სტანდარტების საკალიბრო მრუდიდან, მრუდის ქვედა დიაპაზონში დაბალი სიზუსტის გამო. მხედველობაში მიიღება ექსტრაჰირებისა და გაწმენდის შედეგები. საანგარიშგებო ლიმიტი უნდა დადგინდეს სულ მცირე სამჯერ მეტი, ვიდრე საკონტროლო პროცედურისას. 7.2. ეტალონური ნიმუშების გამოყენება 7.2.1. ეტალონური ნიმუშები უნდა წარმოადგენდეს ნიმუშის მატრიცას, კონგენერულ ნიმუშებს და კონცენტრაციის დიაპაზონს PCDD/F და დიოქსინის მსგავსი PCB-ებისთვის უნდა იყოს მაქსიმალური შემცველობის (ზღვარის) ან მოქმედების ზღურბლის ფარგლებში. 7.2.2. მატრიცის საკონტროლო (სუფთა) და სადაც ეს შეუძლებელია, პროცედურის საკონტროლო (სუფთა) და ეტალონური ნიმუში მაქსიმალურ შემცველობაზე (ზღვარზე) ან მოქმედების ზღურბლზე უნდა იყოს ჩართული თითოეული ტესტის სერიაში. უნდა მოხდეს ამ ნიმუშების ექსტრაჰირება და ტესტირება ერთსა და იმავე დროს იდენტური პირობებით. ეტალონურმა ნიმუშმა უნდა აჩვენოს აშკარად ამაღლებული პასუხი საკონტროლო (სუფთა) ნიმუშთან შედარებით, რაც უზრუნველყოფს ტესტის ვარგისიანობას. ეს ნიმუშები შეიძლება გამოყენებულ იქნეს საკონტროლო და აღდგენის კორექტირებისთვის. 7.2.3. აღდგენის კორექციის შესასრულებლად შერჩეული ეტალონური ნიმუშები უნდა იყოს რეპრეზენტატული გამოსაკვლევი (სატესტო) ნიმუშებისთვის, რაც ნიშნავს, იმას, რომ კონგენერის ნიმუშებმა შეიძლება არ მიგვიყვანოს შემცველობის (ზღვრების) არასათანადო შეფასებამდე. 7.2.4. დამატებითი ეტალონური ნიმუშები მაგ. 0,5x და 2x მაქსიმალური შემცველობის ან მოქმედების ზღურბლზე შეიძლება ჩართული იყოს ტესტის ინტერესის დიაპაზონში სათანადო შესრულების დემონსტრირებისთვის, მაქსიმალური შემცველობის ან მოქმედების ზღურბლის კონტროლისთვის. ერთობლიობაში, ეს ნიმუშები შეიძლება გამოყენებულ იქნეს BEQ ზღვარის გამოსათვლელად გამოსაკვლევ ნიმუშებში (იხ. ამ ქვეთავის პუნქტი 7.1.2.2). 7.3. ზღვრული მნიშვნელობების (cut-off value) განსაზღვრა უნდა დამყარდეს კავშირი BEQ-ში ბიოანალიტიკურ შედეგებსა და TEQ-ში დამადასტურებელ მეთოდით მიღებულ შედეგებს შორის, მაგალითად, მატრიცასთან თავსებადობის საკალიბრო ექსპერიმენტებით, რომლებიც მოიცავს 0, 0,5x, 1x და 2x ML-ზე ეტალონურ ნიმუშებს, თითოეულ დონეზე 6 განმეორებით (n = 24). შესწორების ფაქტორები (საკონტროლო და აღდგენითი) შეიძლება შეფასდეს ამ ურთიერთობიდან, მაგრამ ისინი უნდა შემოწმდეს ამ ქვეთავის 7.2.2-ე პუნქტის შესაბამისად. მაქსიმალური შემცველობის (ზღვარის) ნიმუშის შესაბამისობის ან მოქმედების ზღურბლების კონტროლის შესახებ გადაწყვეტილებისთვის უნდა დადგინდეს ზღვრული (cut-off value) მნიშვნელობები, საჭიროების შემთხვევაში, შესაბამისი მაქსიმალური შემცველობით ან მოქმედების ზღუბლით, რომელიც დადგენილია მხოლოდ PCDD/F-ისა და დიოქსინის მსგავსი PCB-ებისთვის, ან PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამისთვის. ისინი წარმოდგენილია ბიოანალიტიკური შედეგების განაწილების ქვედა საბოლოო-წერტილით (კორექტირებულია საკონტროლოსთვის და აღდგენისთვის), რომელიც შეესაბამება დამადასტურებელი მეთოდის გადაწყვეტილების ზღვარს, რომელიც ეფუძნება 95%-იან სანდოობას, რაც გულისხმობს, რომ ცრუ შესაბამისობის მაჩვენებელი <5%, და RSDR-ზე <25%-ით. დამადასტურებელი მეთოდის გადაწყვეტილების ზღვარი არის მაქსიმალური შემცველობა, განუსაზღვრელობის გაფართოებული განსაზღვრის გათვალისწინებით. ზღვრული (cut-off value) მნიშვნელობა (BEQ-ში) შეიძლება გამოითვალოს ამ ქვეთავის 7.3.1, 7.3.2 და 7.3.3 პუნქტებით განსაზღვრული ერთ-ერთი მიდგომის შესაბამისად (იხ. ამ ქვეთავის სურათი 1). 7.3.1. 95% პროგნოზირების ინტერვალის ქვედა დიაპაზონის (ზოლის) გამოყენება დამადასტურებელი მეთოდის გადაწყვეტილების ზღვარზე:
სადაც:
7.3.2. დადასტურების მეთოდის გადაწყვეტილების ზღვარზე დაბინძურებული ნიმუშების მრავალჯერადი ანალიზის (n ≥ 6) ბიოანალიტიკური შედეგების გაანგარიშება (კორექტირებულია საკონტროლოსთვის და აღდგენისთვის), როგორც მონაცემთა განაწილების ქვედა საბოლოო წერტილი შესაბამისი BEQ-ის მნიშვნელობით:
სადაც: SDR ბიოანალიზის გამოკვლევის შედეგების სტანდარტული გადახრა BEQDL-ზე, რომელიც იზომება ლაბორატორიულ რეპროდუქციულობის (შედეგიანობის) პირობებში. 7.3.3. გაანგარიშება, როგორც ბიოანალიტიკური შედეგების საშუალო მნიშვნელობა (BEQ-ში, კორექტირებულია საკონტროლოსთვის და აღდგენისთვის) ნიმუშების მრავალჯერადი ანალიზის შედეგად (n ≥ 6), რომლებიც მაქსიმალური შემცველობის ან მოქმედების ზღურბლის ორ მესამედზეა დაბინძურებული და რომ ეს ზღვარი, დაკვირვების საფუძველზე, იქნება დაახლოებით ზღვრული (cut-off value) მნიშვნელობა, განსაზღვრული ამ ქვეთავის 7.3.1-ე ან 7.3.2-ე პუნქტების შესაბამისად: ზღვრული მნიშვნელობების (cut-off value) გაანგარიშება 95%-იანი სანდობის საფუძველზე, რაც გულისხმობს, რომ ცრუ შესაბამისობის მაჩვენებელი <5% და RSDR <25%: – 95%-იანი პროგნოზირების ინტერვალის ქვედა დიაპაზონიდან (ზოლიდან) დამადასტურებელი მეთოდის გადაწყვეტილების ზღვარზე. – ნიმუშების მრავალჯერადი გამოკვლევიდან (n≥6), რომელიც დაბინძურებულია დამადასტურებელი მეთოდის გადაწყვეტილების ზღვარზე, როგორც მონაცემთა განაწილების ქვედა ბოლო-წერტილი (ნახაზზე გამოსახულია ზარის ფორმის მრუდი) შესაბამისი საშუალო BEQ მნიშვნელობით.
7.3.4. შეზღუდვები ზღვრულ (cut-off value) მნიშვნელობებზე ვალიდაციის დროს მიღებული BEQ-ზე დაფუძნებული ზღვრული (cut-off value) მნიშვნელობები (რომელიც გაანგარიშებულია RSDR-დან), ნიმუშების შეზღუდული რაოდენობის გამოყენებით სხვადასხვა მატრიცული/კონგენერული შაბლონით, შეიძლება უფრო მაღალი იყოს, ვიდრე TEQ-ზე დაფუძნებული მაქსიმალური შემცველობა ან მოქმედების ზღურბლები, მაღალი სიზუსტის გამო, ვიდრე ეს მიღწევადია რუტინაში, როდესაც კონგენერების შაბლონების უცნობი სპექტრი უნდა გაკონტროლდეს. ასეთ შემთხვევებში, ზღვრული (cut-off value) მნიშვნელობები უნდა გამოითვალოს RSDR = 25%-ით, ან უპირატესობა უნდა მიენიჭოს მაქსიმალურ შემცველობას ან მოქმედების ზღურბლის ორ მესამედს. 7.4. შესრულების მახასიათებლები 7.4.1. ვინაიდან ბიოანალიტიკურ მეთოდებში შიდა სტანდარტების გამოყენება არ შეიძლება, ბიოანალიტიკური მეთოდების განმეორებადობის შესახებ ტესტები უნდა ჩატარდეს ტესტის სერიებში და მათ შორის სტანდარტული გადახრის შესახებ ინფორმაციის მისაღებად. განმეორებადობა უნდა იყოს 20%-ზე ნაკლები, ხოლო ლაბორატორიული რეპროდუქციულობის უნარი 25%-ზე ნაკლები. ეს უნდა დაეფუძნოს BEQ-ში გამოანგარიშებულ ზღვარს საკონტროლო და აღდგენითი შესწორების შემდეგ. 7.4.2. ვალიდაციის პროცესის ფარგლებში, ნაჩვენები უნდა იყოს ტესტი, საკონტროლო ნიმუშსა და ზღვრულ (cut-off value) მნიშვნელობას შორის განსასხვავებლად, რაც საშუალებას იძლევა მოვახდინოთ შესაბამისი ზღვრული (cut-off value) მნიშვნელობაზე მაღალი შედეგების მქონე ნიმუშების იდენტიფიცირება (იხ. ამ ქვეთავის პუნქტი 7.1.2). 7.4.3. საჭიროა განისაზღვროს სამიზნე ნაერთები, შესაძლო შემცვლელი ნივთიერებები და მაქსიმალურად დასაშვები საკონტროლო ზღვრები. 7.4.4. პასუხში არსებული სტანდარტული გადახრა ან კონცენტრაცია, რომელიც გამოითვლება ნიმუშის ექსტრაქტის სამგზის განსაზღვრის პასუხიდან (შესაძლებელია მხოლოდ სამუშაო დიაპაზონში), არ შეიძლება იყოს 15% -ზე მეტი. 7.4.5. BEQ-ში (საკონტროლო და მაქსიმალურ შემცველობაზე ან მოქმედების ზღურბლზე) გამოხატული ეტალონური ნიმუშის (ნიმუშების) შესწორებული შედეგები გამოყენებული უნდა იქნეს ბიოანალიტიკური მეთოდის მუშაობის შესაფასებლად მუდმივი დროის განმავლობაში. 7.4.6. ხარისხის კონტროლის დიაგრამები კონტროლის პროცედურისთვის და თითოეული ტიპის ეტალონური ნიმუში უნდა იქნეს ჩაწერილი და შემოწმდეს, რათა დავრწმუნდეთ, რომ ანალიტიკური შესრულება(გამოკვლევა) შეესაბამება მოთხოვნებს, კერძოდ, კონტროლის პროცედურისთვის, მოთხოვნილი მინიმალური სხვაობის გათვალისწინებით სამუშაო დიაპაზონის ქვედა ბოლოსთან დაკავშირებით, და ეტალონური ნიმუშებისთვის, შიდალაბორატორიული რეპროდუქციულობასთან დაკავშირებით. კონტროლის პროცედურა უნდა კონტროლდეს ისე, რომ თავიდან ავიცილოთ ცრუ-შესაბამისობის შედეგები გამოკლებისას. 7.4.7. საეჭვო ნიმუშების დამადასტურებელი მეთოდებისა და შესაბამისი ნიმუშების (მინიმუმ 20 ნიმუში თითო მატრიცაზე) 2%-დან 10%-ის შედეგები უნდა შეგროვდეს და გამოყენებულ იქნეს სკრინინგის მეთოდის მუშაობისა და BEQ-სა და TEQ-ს შორის კავშირის შესაფასებლად. ეს მონაცემთა ბაზა შეიძლება გამოყენებულ იქნეს ზღვრული (cut-off value) მნიშვნელობების ხელახალი შეფასებისთვის, რომლებიც გამოიყენება რუტინული ნიმუშების დადასტურებული (ვალიდირებული) მატრიცებისთვის. 7.4.8. მეთოდის წარმატებული მუშაობის ჩვენება ასევე შესაძლებელია რგოლურ გამოცდაში მონაწილეობით. რგოლურ გამოცდაში გამოკვლეული ნიმუშების შედეგები, რომლებიც მოიცავს კონცენტრაციის დიაპაზონს, მაგ.: 2 × მაქსიმალურ ზღვრამდე, შეიძლება მოიცავდეს ცრუ შესაბამისობის მაჩვენებლის შეფასებას, თუ ლაბორატორიას შეუძლია აჩვენოს მისი წარმატებული შესრულება. ნიმუშები მოიცავს ყველაზე ხშირად კონგენერის ნიმუშებს, რომლებიც წარმოადგენენ სხვადასხვა წყაროს. 7.4.9. ინციდენტების დროს, ზღვრული (cut-off value) მნიშვნელობები შეიძლება ხელახლა შეფასდეს, რაც ამავდროულად ასახავს ამ კონკრეტული ინციდენტის სპეციფიკურ მატრიცას და კონგენერის ნიმუშებს.
8. შედეგების ანგარიშგება 8.1. დამადასტურებელი მეთოდები 8.1.1. გამოკვლევის შედეგები უნდა შეიცავდეს ინდივიდუალური PCDD/F შემცველობას და დიოქსინის მსგავსი PCB კონგენერების შემცველობას და TEQ-მნიშვნელობები უნდა იყოს მოხსენიებული, როგორც ქვედა, ზედა და საშუალო ზღვარი, რათა შედეგების შესახებ შეტანილ იქნეს მაქსიმალური ინფორმაცია ანგარიშში, და ამგვარად, შესაძლებელი გახდეს შედეგების ინტერპრეტაცია კონკრეტული მოთხოვნების შესაბამისად. 8.1.2. ანგარიში უნდა მოიცავდეს მეთოდს, რომელიც გამოიყენება PCDD/F და დიოქსინის მსგავსი PCB-ების ექსტრაჰირებისთვის. 8.1.3. ინდივიდუალური შიდა სტანდარტების აღდგენა ხელმისაწვდომი უნდა იყოს იმ შემთხვევაში, თუ აღდგენა ამ ქვეთავის 6.2.5-ე პუნქტში მითითებული დიაპაზონის მიღმაა, მაქსიმალური შემცველობის გადაჭარბების შემთხვევაში (ამ შემთხვევაში, წარმოებს ორიდან ერთის აღდგენა განმეორებითი (დუბლიკატი) გამოკვლევით) და სხვა შემთხვევაში, მოთხოვნისას. 8.1.4. რადგან ნიმუშის შესაბამისობის საკითხის გადაწყვეტისას მხედველობაში უნდა იქნეს მიღებული განუსაზღვრელობის გაფართოებული განსაზღვრა, ეს პარამეტრი უნდა იყოს ხელმისაწვდომი. ამრიგად, გამოკვლევის შედეგები უნდა აღინიშნოს, როგორც x +/– U, სადაც x არის გამოკვლევის შედეგი და U არის განუსაზღვრელობის გაფართოებული განსაზღვრა დაფარვის 2 ფაქტორის გამოყენებით, რომელიც იძლევა დაახლოებით 95%-იან სანდოობის დონეს. PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ცალ-ცალკე განსაზღვრის შემთხვევაში, PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ცალკეული გამოკვლევის შედეგების სავარაუდო გაფართოებული განუსაზღვრელობის ჯამი, გამოყენებული უნდა იქნეს PCDD/F და დიოქსინის მსგავსი PCB-ის ჯამებისთვის. 8.1.5. შედეგები უნდა აისახოს იმავე ერთეულებში და სულ მცირე იმავე რაოდენობის მნიშვნელოვან ციფრებში, როგორც საქართველოს კანონმდებლობით დადგენილი მაქსიმალური ზღვრებია. 8.2. ბიოანალიტიკური სკრინინგის მეთოდები 8.2.1. სკრინინგის შედეგი გამოიხატება, როგორც „შესაბამისი“ ან „საეჭვოა, რომ არ არის შესაბამისი“ („საეჭვო“). 8.2.2. გარდა ამისა, BEQ-ით და არა TEQ-ით გამოხატული მითითებული შედეგი PCDD/F და/ან დიოქსინის მსგავსი PCB-ებისთვის, შეიძლება არ იყოს მოცემული. 8.2.3. ნიმუშები, რომელზეც პასუხი საანგარიშგებო ზღვრის ქვემოთაა, უნდა გამოიხატოს, როგორც ”საანგარიშო ზღვარზე დაბალი”. სამუშაო დიაპაზონის ზემოთ პასუხის მქონე ნიმუშები უნდა ჩაიწეროს, როგორც „აჭარბებს სამუშაო დიაპაზონს“ და სამუშაო დიაპაზონის ზედა ბოლოს შესაბამისი დონე უნდა იყოს მოცემული BEQ-ში. 8.2.4. თითოეული ტიპის ნიმუშის მატრიცისთვის, ანგარიშში უნდა აღინიშნოს მაქსიმალური შემცველობა ან მოქმედების ზღურბლი, რომელსაც ემყარება შეფასება. 8.2.5. ანგარიშში უნდა აღინიშნოს გამოყენებული ტესტის ტიპი, ტესტის ძირითადი პრინციპი და საკალიბრო სახე. 8.2.6. ანგარიშში უნდა აღინიშნოს გამოყენებული მეთოდი, რომლითაც მოხდა PCDD/F და დიოქსინის მსგავსი PCB-ების ექსტრაჰირება. 8.2.7. იმ შემთხვევაში, თუ ნიმუშები საეჭვოა, რომ შეუსაბამოა, ანგარიშში უნდა აღინიშნოს ჩასატარებელი მოქმედებების შესახებ. PCDD/F-ების კონცენტრაცია და PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამი მომატებული შემცველობის ნიმუშებში უნდა დადასტურდეს დამადასტურებელი მეთოდით. 8.2.8. შეუსაბამო შედეგები, უნდა აღინიშნოს ანგარიშში, მხოლოდ დამადასტურებელი გამოკვლევიდან. 8.3. ფიზიკურ-ქიმიური სკრინინგის მეთოდები 8.3.1. სკრინინგის შედეგი გამოიხატება, როგორც „შესაბამისი“ ან „საეჭვოა, რომ არ არის შესაბამისი“ („საეჭვო“). 8.3.2. თითოეული ტიპის ნიმუშის მატრიცისთვის, ანგარიშში უნდა აღინიშნოს მაქსიმალური შემცველობა ან მოქმედების ზღურბლი, რომელსაც ემყარება შეფასება. 8.3.3. გარდა ამისა, ზღვრები ინდივიდუალური PCDD/F ან/და დიოქსინის მსგავსი PCB კონგენერების და TEQ მნიშვნელობებისთვის, ანგარიშში შეიძლება აღინიშნოს, როგორც ქვედა, ზედა და საშუალო ზღვარი. შედეგები უნდა აისახოს იმავე ერთეულებში და სულ მცირე იმავე რაოდენობის მნიშვნელოვან ციფრებში, როგორც საქართველოს კანონმდებლობით დადგენილი მაქსიმალური ზღვრებია. 8.3.4. ინდივიდუალური შიდა სტანდარტების აღდგენა ხელმისაწვდომი უნდა იყოს იმ შემთხვევაში, თუ აღდგენა ამ ქვეთავის 6.2.5-ე პუნქტში მითითებული დიაპაზონის მიღმაა, მაქსიმალური შემცველობის გადაჭარბების შემთხვევაში (ამ შემთხვევაში, წარმოებს ორიდან ერთის აღდგენა განმეორებითი (დუბლიკატი) გამოკვლევით) და სხვა შემთხვევაში, მოთხოვნისას. 8.3.5. ანგარიშში უნდა აღინიშნოს GC-MS-ის გამოყენებული მეთოდი. 8.3.6. ანგარიშში უნდა აღინიშნოს გამოყენებული მეთოდი, რომლითაც მოხდა PCDD/F და დიოქსინის მსგავსი PCB-ების ექსტრაჰირება. 8.3.7. იმ შემთხვევაში, თუ ნიმუშები საეჭვოა, რომ შეუსაბამოა, ანგარიშში უნდა აღინიშნოს ჩასატარებელი მოქმედებების შესახებ. PCDD/F-ების კონცენტრაცია და PCDD/F-ებისა და დიოქსინის მსგავსი PCB-ების ჯამი მომატებული ზღვრების მქონე ნიმუშებში უნდა დადასტურდეს დამადასტურებელი მეთოდით. 8.3.8. შეუსაბამობის დადგენა შესაძლებელია მხოლოდ დამადასტურებელი გამოკვლევის შემდეგ. ქვეთავი III ცხოველის საკვებში არადიოქსინის მსგავსი PCB-ების შემცველობის სახელმწიფო კონტროლისათვის ნიმუშის მომზადებასა და გამოკვლევის მეთოდებთან დაკავშირებული მოთხოვნები 1. გამოყენების სფერო ამ თავში მოცემული მოთხოვნები გამოყენებული უნდა იქნეს, როდესაც ცხოველის საკვების გამოკვლევა ხდება არადიოქსინის მსგავსი PCB-ების შემცველობის სახელმწიფო კონტროლისთვის, და სხვა მარეგულირებელი მიზნებისათვის, ნიმუშის მომზადებისა და გამოკვლევის მოთხოვნების გათვალისწინებით, რაც მოიცავს ცხოველის საკვების ბიზნესოპერატორის მიერ კონტროლს, რათა უზრუნველყოს „ცხოველის საკვების ჰიგიენის წესის დამტკიცების შესახებ“ – საქართველოს მთავრობის 2020 წლის 13 მარტის N173 დადგენილებით განსაზღვრული მოთხოვნების შესრულება. 2. გამოსაყენებელი გამოვლენის მეთოდები გაზის ქრომატოგრაფია/ელექტრონების დაჭერის გამოვლენა (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS ან ეკვივალენტური მეთოდები. 3. საინტერესო ანალიტების იდენტიფიკაცია და დადასტურება 3.1. ფარდობითი შეკავების დრო შიდა სტანდარტებთან ან ეტალონურ სტანდარტებთან დაკავშირებით (დასაშვები გადახრა +/– 0,25%). 3.2. არადიოქსინის მსგავსი PCB-ების გაზ-ქრომატოგრაფიული გამიჯვნა ხელშემშლელი ნივთიერებებისაგან, განსაკუთრებით თანაელუირებული PCB-ებისაგან, განსაკუთრებით იმ შემთხვევაში, თუ ნიმუშების შემცველობა (ზღვრები) სამართლებრივ ფარგლებშია და როდესაც შეუსაბამობა დასადასტურებელია20. 3.3. GC-MS ტექნიკის მოთხოვნები მონიტორინგი სულ მცირე მოლეკულური იონების ან დამახასიათებელი იონების შემდეგ რაოდენობაზე: ა) ორი კონკრეტული იონი HRMS-სთვის; ბ) სამი სპეციფიკური იონი LRMS-ისთვის; გ) ორი სპეციფიური პრეკურსორი (წინამორბედი) იონი, რომლებსაც ინდივიდუალურად გააჩნიათ ერთი სპეციფიური შესაბამისი გარდამავალი პროდუქტის იონი MS-MS-სთვის. მაქსიმალური დასაშვები ტოლერანტობა ფარდობითი შემცველობის შერჩეული მასის ფრაგმენტებისთვის: შერჩეული მას-ფრაგმენტების ფარდობითი შემცველობის გადახრა თეორიული ფარდობითი შემცველობის ან საკალიბრო სტანდარტიდან სამიზნე იონისთვის (მონიტორინგს ექვემდებარება ყველაზე მეტად გავრცელებული იონი) და შერჩეული იონისთვის: ± 15%. 3.4. GC-ECD ტექნიკის მოთხოვნები შედეგები, რომლებიც აღემატება მაქსიმალურ ზღვარს უნდა დადასტურდეს სხვადასხვა პოლარობის სტაციონარული ფაზების მქონე ორი GC სვეტით. 4. მეთოდის შესრულების დემონსტრირება მეთოდის შესრულება ვალიდირებული უნდა იქნეს მაქსიმალური შემცველობის (ზღვარის) დიაპაზონში (0,5-დან 2-ჯერ მაქსიმალურ ზღვარამდე) განმეორებითი გამოკვლევისათვის ვარიაციის მისაღები კოეფიციენტით (იხილეთ შუალედური სიზუსტის მოთხოვნები, ამ ქვეთავის მე-9 პუნქტში). 5. რაოდენობრივი შეზღუდვა არადიოქსინის მსგავსი PCB-ების LOQ-ების21 ჯამი არ უნდა იყოს მაქსიმალური შემცველობის მესამედზე მეტი22. 6. ხარისხის კონტროლი რეგულარული ბლანკების (ბრმა) კონტროლი, დანამატებიანი ნიმუშების (spiked sample) გამოკვლევა, ხარისხის კონტროლის ნიმუშები, ლაბორატორიათაშორის კვლევებში მონაწილეობა შესაბამის მატრიცების გამოყენებით. 7. აღდგენის კონტროლი 7.1. შესაფერისი შიდა სტანდარტები ფიზიკურ-ქიმიური მახასიათებლებით გამოყენებული უნდა იქნეს ინტერეს-ანალიტებთან შესადარებლად. 7.2. შიდა სტანდარტების დამატება: პროდუქტებზე დამატება (ესტრაჰირებამდე და გაწმენდის პროცესამდე). 7.3. მოთხოვნები მეთოდებისთვის, რომელიც იყენებს ექვსივე იზოტოპნიშანდებულ არადიოქსინის მსგავსი PCB-ის კონგენერებს: ა) შედეგები უნდა შესწორდეს შიდა სტანდარტების აღსადგენად; ბ) იზოტოპნიშანდებული შიდა სტანდარტების აღდგენა უნდა იყოს 60 და 120%; გ) დასაშვებია ცალკეული კონგენერებისთვის, აღდგენის დაბალი ან მაღალი მაჩვენებლები, რომელთა კონტრიბუცია არადიოქსინის მსგავსი PCB-ების ჯამში 10%-ზე ნაკლებია. 7.4. მოთხოვნები მეთოდებისთვის, რომლებიც არ იყენებენ ყველა ექვს იზოტოპნიშანდებულ შიდა სტანდარტებს, ან სხვა შიდა სტანდარტებს: ა) შიდა სტანდარტის (სტანდარტების) აღდგენა უნდა გაკონტროლდეს თითოეული ნიმუშისთვის; ბ) შიდა სტანდარტის (სტანდარტების) აღდგენა უნდა იყოს 60-დან 120%-მდე; გ) შედეგები უნდა შესწორდეს შიდა სტანდარტების აღდგენის გამოყენებით. 7.5. არანიშანდებული კონგენერების აღდგენა უნდა შემოწმდეს დანამატებიანი ნიმუშებით (spiked sample) ან ხარისხის კონტროლის ნიმუშებით, რომელთა კონცენტრაცია მაქსიმალური შემცველობის ფარგლებშია. ამ კონგენერების აღდგენები უნდა ჩაითვალოს მისაღებად, თუ ისინი 60-დან 120%-მდეა. 8. მოთხოვნები ლაბორატორიებისთვის ლაბორატორიები უნდა იყვნენ აკრედიტებული აღიარებული ორგანოს მიერ, რომლებიც მუშაობენ ISO-ს 58-ე სახელმძღვანელოს შესაბამისად, იმაში დასარწმუნებლად, რომ ისინი იყენებენ ანალიტიკური ხარისხის უზრუნველყოფას. ლაბორატორიები აკრედიტებული უნდა იყვნენ EN ISO/IEC 17025 სტანდარტის შესაბამისად. გარდა ამისა, საჭიროების შემთხვევაში, დაცული უნდა იქნეს პრინციპები, როგორც ეს აღწერილია ტექნიკურ სახელმძღვანელოში განუსაზღვრელობის განსაზღვრის შეფასებისა და PCB ანალიზის რაოდენობრივი შეფასების ლიმიტებისთვის.23 9. შესრულების მახასიათებლები: კრიტერიუმები არადიოქსინის მსგავსი PCB-ების ჯამისთვის მაქსიმალურ შემცველობაზე (ზღვარზე)
10. შედეგების ანგარიშგება 10.1. გამოკვლევის შედეგები უნდა შეიცავდეს ინდივიდუალური არადიოქსინის მსგავსი PCB-ების შემცველობას (ზღვარს) და PCB კონგენერების ჯამს, რომლებიც აღინიშნა, როგორც ქვედა, ზედა და საშუალო ზღვარი, რათა შედეგების შესახებ შეტანილ იქნეს მაქსიმალური ინფორმაცია ანგარიშში, და ასე, შესაძლებელი გახდეს შედეგების ინტერპრეტაცია კონკრეტული მოთხოვნების შესაბამისად. 10.2. ანგარიში უნდა მოიცავდეს მეთოდს, რომელიც გამოიყენება PCB-ების ექსტრაჰირებისთვის. 10.3. ინდივიდუალური შიდა სტანდარტების აღდგენა ხელმისაწვდომი უნდა იყოს იმ შემთხვევაში, თუ აღდგენები ამ ქვეთავის მე-7 პუნქტში მითითებულ დიაპაზონის მიღმაა, მაქსიმალური ზღვარის გადაჭარბების შემთხვევაში და სხვა შემთხვევაში, მოთხოვნისას. 10.4. რადგან ნიმუშის შესაბამისობის საკითხის გადაწყვეტისას მხედველობაში უნდა იქნეს მიღებული განუსაზღვრელობის გაფართოებული განსაზღვრა, ეს პარამეტრი უნდა იყოს ხელმისაწვდომი. ამრიგად, გამოკვლევის შედეგები უნდა აღინიშნოს, როგორც x +/– U, სადაც x არის გამოკვლევის შედეგი და U არის განუსაზღვრელობის გაფართოებული განსაზღვრა დაფარვის 2 ფაქტორის გამოყენებით, რომელიც იძლევა დაახლოებით 95%-იან სანდოობას. 10.5. შედეგები უნდა აისახოს იმავე ერთეულებში და სულ მცირე იმავე რაოდენობის მნიშვნელოვან ციფრებში, როგორც საქართველოს კანონმდებლობით დადგენილი მაქსიმალური ზღვრებია.
დანართი № 6
ცხოველის საკვების კონტროლისას ცხოველური წარმოშობის შემადგენელი ნაწილაკების განსაზღვრასთან დაკავშირებული გამოკვლევის მეთოდები
1. მიზანი და გამოყენების სფერო ცხოველის საკვებთან დაკავშირებული ცხოველური წარმოშობის შემადგენელი ნაწილაკების განსაზღვრა ხორციელდება სინათლის მიკროსკოპით ან პოლიმერაზული ჯაჭვური რეაქციით (შემდეგში – PCR) ამ დანართში მითითებული დებულებების შესაბამისად. ამ ორი მეთოდით შესაძლებელია განხორციელდეს ცხოველური წარმოშობის შემადგენელი ნაწილაკების გამოვლენა ცხოველის საკვებ მასალასა და ცხოველის კომბინირებულ საკვებში. თუმცა ეს მეთოდები არ იძლევა იმის საშუალებას, რომ განხორციელდეს ცხოველის საკვებ მასალასა და ცხოველის კომბინირებულ საკვებში ცხოველური წარმოშობის შემადგენელი ნაწილაკების რაოდენობრივი დადგენა. ორივე მეთოდს აქვს გამოვლენის ზღვარი 0.1%-ზე (წ/წ) ქვევით. PCR მეთოდით შესაძლებელია ცხოველის საკვებ მასალასა და ცხოველის კომბინირებულ საკვებში ცხოველური წარმოშობის შემადგენელი ტაქსონომიური ჯგუფების იდენტიფიცირება. ეს მეთოდები გამოიყენება „ტექნიკური რეგლამენტი – ზოგიერთი გადამდები ღრუბლისებრი ენცეფალოპათიების პრევენციის, კონტროლისა და აღმოფხვრის წესის დამტკიცების შესახებ“ -საქართველოს მთავრობის 2016 წლის 28 დეკემბრის №600 დადგენილების მე-8 მუხლის პირველი პუნქტითა და თავი IX-ით და „ტექნიკური რეგლამენტის – „ცხოველური წარმოშობის არასასურსათო დანიშნულების პროდუქტისა (მათ შორის, ცხოველური ნარჩენების) და მეორეული პროდუქტის, რომლებიც არ არის გამიზნული ადამიანის მიერ მოხმარებისათვის, ჯანმრთელობისა და ამ საქმიანობასთან დაკავშირებული ბიზნესოპერატორის აღიარების წესების“ დამტკიცების შესახებ“ – საქართველოს მთავრობის 2017 წლის 29 დეკემბრის №605 დადგენილების მე-10 მუხლის პირველი პუნქტით გათვალისწინებული აკრძალვების საკონტროლოდ. ცხოველის საკვების ტიპის შემოწმების გათვალისწინებით, ეს მეთოდები შესაძლებელია გამოყენებულ იქნეს ერთი სამუშაო პროტოკოლის ფარგლებში, ცალკე ან კომბინირებულად სტანდარტული სამუშაო ინსტრუქციის შესაბამისად (SOP), რომელიც დადგენილია ცხოველის საკვებში ცხოველური ცილების განმსაზღვრელ (შემდეგში – EURL-AP) ევროკავშირის – რეფერენს ლაბორატორიის მიერ და გამოქვეყნებულია მის ვებგვერდზე (http://eurl.craw.eu//).
2. მეთოდები 2.1. სინათლის მიკროსკოპია 2.1.1. გამოყენების პრინციპი სინათლის მიკროსკოპიის პრინციპია გამოკვლევისათვის გაგზავნილი ცხოველური წარმოშობის შემადგენელი ნაწილების, რომლებიც შესაძლოა წარმოდგენილი იყოს ცხოველის საკვებ მასალაში და კომბინირებულ საკვებში, იდენტიფიცირება ტიპური და მიკროსკოპულად იდენტიფიცირებადი მახასიათებლების საფუძველზე, როგორიცაა კუნთოვანი ბოჭკო და ხორცის სხვა ნაწილაკები, ხრტილი, ძვლები, რქა, ბალანი, ჯაგარი, სისხლი, რძის ბურთულები, ლაქტოზას კრისტალები, ბუმბული, კვერცხის ნაჭუჭი, თევზის ფხა და ქერცლი. 2.1.2. რეაგენტები და აღჭურვილობა 2.1.2.1. რეაგენტები 2.1.2.1.1. კონცენტრირებული აგენტი 2.1.2.1.1.1. ტეტრაქლორეთილენის (კუთვნილი მასა 1,62) 2.1.2.1.2. მღებავი რეაგენტი 2.1.2.1.2.1. ალიზარინის წითელი ხსნარი (განაზავეთ 2.5 მლ 1 მოლი მარილმჟავა 100 მლ წყალში და ხსნარს დაუმატეთ 200 მგ ალიზარინის წითელი); 2.1.2.1.3. საფიქსაციო ნიადაგი (mounting medium): 2.1.2.1.3.1. ტუტე ხსნარი (NaOH 2,5% w/v ან KOH 2,5% w/v) 2.1.2.1.3.2. გლიცეროლი (განუზავებული, სიბლანტე: 1,490 cP) ან საფიქსაციო ნიადაგი (mounting medium) ეკვივალენტური თვისებებით არამუდმივი სლაიდის მოსამზადებლად. 2.1.2.1.3.3. Norland® ოპტიკური წებო 65 (სიბლანტე 1200 სანტიპაუზი) ან ფისი ეკვივალენტური მახასიათებლებით მუდმივი პრეპარატის დამზადებისთვის; 2.1.2.1.4. საფიქსაციო ხსნარი (mounting medium) მღებავი მახასიათებლებით: 2.1.2.1.4.1. ლუგოლის ხსნარი (გახსენით 2გრ კალიუმის იოდატი 100 მლ წყალში და დაამატეთ 1გ იოდი და შეანჯღრიეთ); 2.1.2.1.4.2. ცისტინის რეაგენტი (1 გ ტყვიის აცეტატი, 10 გ NaOH/100 მლ წყალი); 2.1.2.1.4.3. ფელინგის რეაგენტი (მზადდება გამოყენებამდე A და B ხსნარების თანაბარი (1/1) რაოდენობებისაგან. ხსნარი A: 6.9 გ. სპილენძის(II) სულფატის პენტაჰიდრატი გახსენით 100 მლ წყალში. ხსნარი B: 34.6 გ კალიუმ-ნატრიუმის ტარტრატის ტეტრაჰიდრატი და 12 გ NaOH გახსენით 100მლ წყალში); 2.1.2.1.4.4. ტეტრამეთილბენზიდინი/წყალბადის ზეჟანგი (გახსენით 1გ 3,3’,5,5’ ტეტრამეთილბენზიდინი (TMB) 100 მლ დაკრისტალებულ ძმარმჟავაში და 150 მლ წყალში. გამოყენებამდე, აურიეთ TMB-ის ხსნარის 4 წილი 3%-იანი წყალბადის ზეჟანგის 1 წილთან. 2.1.2.1.5. სინათლის მიკროსკოპიისათვის საჭირო გამრეცხი აგენტები: 2.1.2.1.5.1. ეთანოლი > = 96% (ტექნიკური); 2.1.2.1.5.2. აცეტონი (ტექნიკური); 2.1.2.1.6. მათეთრებელი რეაგენტი – კომერციული ნატრიუმ ჰიპოქლორიდის ხსნარი (9 -14 %-იანი აქტიური ქლორი); 2.1.2.1.6.1. ნატრიუმის ჰიპოქლორიტის კომერციული ხსნარი (9 – 14% აქტიური ქლორი). 2.1.2.2. სინათლის მიკროსკოპიისათვის საჭირო აღჭურვილობა: 2.1.2.2.1. ანალიზური სასწორი 0.001 გ-იანი სიზუსტით; 2.1.2.2.2. დამფქვავი/დამაქუცმაცებელი მოწყობილობა: წისქვილი ან როდინი; როტორული წისქვილის გამოყენების შემთხვევაში აკრძალულია წისქვილის საცრები ≤ 0,5 მმ. 2.1.2.2.3. საცერი კვადრატული ბადით 0.25მმ და 1მმ სიგანის; ნიმუშის წინასწარი გაცრის გარდა, საცრების დიამეტრი არ უნდა აღემატებოდეს 10 სმ-ს, რათა თავიდან იქნეს აცილებული მასალების დაკარგვა. საცრების დაკალიბრება არ არის საჭირო. 2.1.2.2.4. 250 მლ ტევადობის კონუსის ფორმის მინის გამყოფი ძაბრი ტეფლონის (პოლიტეტრაფთორეთანი) ან მინის ონკანით კონუსის ძირში. ონკანის გამხსნელის დიამეტრი უნდა იყოს ≥4მმ. ან, კონუსის ძირის მქონე მენზურა შესაძლებელია იქნეს გამოყენებული იმ პირობით, თუ ლაბორატორიამ აჩვენა რომ გამოვლენის დონე არის კონუსის ფორმის მქონე მინის ძაბრის ეკვივალენტური;
2.1.2.2.5. სტერეომიკროსკოპია, რომელიც მოიცავს სულ მცირე 6.5-დან 40-მდე მაქსიმალური გადიდების დიაპაზონს; 2.1.2.2.6. მრავალფუნქციური მიკროსკოპია, რომელიც მოიცავს სულ მცირე 100-დან 400-მდე მაქსიმალური (საბოლოო) გადიდების დიაპაზონს ტრანსმისიული სინათლის ნათელი ველით. პოლარიზებული სინათლე და დიფერენცირებული ინტერფერენციული კონტრასტი შესაძლებელია გამოყენებულ იქნეს დამატებით; 2.1.2.2.7. სტანდარტული ლაბორატორიული მინის ჭურჭელი; 2.1.2.2.8. მოწყობილობა სლაიდების მომზადებისთვის: კლასიკური მიკროსკოპული სლაიდები, ღრუს მქონე სლაიდები, საფარი მინები (20×20 მმ), პინცეტები, წვრილი შპადელი. 2.1.2.2.9. ლაბორატორიული ღუმელი 2.1.2.2.10. ცენტრიფუგა 2.1.2.2.11. ფილტრის ქაღალდი: ცელულოზის ხარისხობრივი ფილტრი (ფორების ზომა 4-11 მკმ) 2.1.3. შერჩევა და ნიმუშის მომზადება 2.1.3.1. ნიმუშის აღება 2.1.3.1. გამოყენებული იქნება ამ წესის დანართი №1-ით განსაზღვრული მოთხოვნების შესაბამისად აღებული რეპრეზენტატიული ნიმუში. 2.1.3.2. სიფრთხილის ზომები ლაბორატორიული ჯვარედინი დაბინძურების თავიდან ასაცილებლად, ყველა მრავალჯერადად გამოყენებადი მოწყობილობა გაწმენდილი უნდა იქნეს გამოყენებამდე. ძაბრის ნაწილები უნდა დაიშალოს გაწმენდამდე. ძაბრის ნაწილები და მინის ჭურჭელი უნდა გაირეცხოს წინასწარ ხელით და შემდეგ სარეცხ მანქანაში. საცერი გაწმენდილი უნდა იქნეს სინთეტიკური მაგარ ბეწვიანი ჯაგრისით. საცერის საბოლოო წმენდა აცეტონითა და კომპრესირებული ჰაერით არის რეკომენდირებული მას შემდეგ, როცა განხორციელდება ისეთი ცხიმოვანი ქსოვლის გაცრა როგორიცაა თევზის ფქვილი. 2.1.3.3. ზეთისა და ქონისგან განსხვავებული ნიმუშების მომზადებისას: 2.1.3.3.1. ნიმუშის გაშრობა: ნიმუში, რომლის სინესტის შემცველობა >14%-ზე, დამუშავებამდე უნდა იქნეს გამომშრალი; 2.1.3.3.2. ნიმუშიის წინასწარი გაცრა: რეკომენდებულია წინასწარ გაიცრას 1მმ ცხოველის გრანულირებული (პალეტირებული) საკვები და მარცვალი იმისთვის, რომ საბოლოოდ მომზადდეს და გამოკვლევა ჩაუტარდეს და ცალკე განხორიელდეს ანგარიშგება შედეგად მიღებულ ორი ფრაქციაზე, რომლებიც მიჩნეული უნდა იქნეს, როგორც განსხვავებული ნიმუშები; 2.1.3.3.3. ქვენიმუშის აღება და დაფქვა: გამოკვლევისათვის და საბოლოოდ დასაფქველად სულ მცირე 50 გრამი ნიმუში უნდა იქნეს ქვენიმუშისთვის გამოყენებული; 2.1.3.3.4. ნალექის ექსტრაჰირება და მომზადება: დაფქული ქვენიმუშის პორცია – 10 გ (0.01გ-მდე სიზუსტით) ჩაეშვება ძაბრში ან კონუსის ძირის მქონე მენზურაში და 50 მლ ტეტრაქლოროეთილენი დაემატება. ძაბრში ჩაშვებული პორცია უნდა შემოიფარგლოს 3 გ-მდე 10%-ზე მეტ ნალექის წარმომქმნელი თევზის ფქვილის ან სხვა სუფთა ცხოველური პროდუქტების, მინერალური ინგრედიენტების ან პრემიქსების შემთხვევაში. მიქსტურა უნდა იქნეს ძლიერად შენჯღრეული სულ მცირე 30 წამით და მიკრული ნაწილაკების მოცილების მიზნით უნდა დაემატოს ფრთხილად სულ მცირე 50 მლ-ით მეტი ტეტრაქლორეთილენი ვიდრე ირეცხება ძაბრის შიდა ზედაპირი. მიღებული მიქსტურა უნდა გაჩერდეს სულ მცირე 5 წუთი, ვიდრე ნალექი არ გამოცალკევდება ონკანის გახსნის შედეგად; თუ გამოყენებულია კონუსის ძირის მქონე მენზურა, მაშინ მიქსტურა უნდა აირიოს ძლიერად სულ მცირე 15 წამი და მენზურას გვერდებზე მიკრული ნებისმიერი ნაწილაკი ფრთხილად უნდა ჩამოირეცხოს სულ მცირე 10 მლ სუფთა ტეტრაქლორეთილენით შიდა ზედაპირზე. მიქსტურა უნდა გაჩერდეს 3 წუთი და შემდეგ ხელახლა შეინჯღრეს 15 წამი და მენზურას გვერდებზე მიკრული ნებისმიერი ნაწილაკი გვერდზე ფრთხილად უნდა იქნეს ჩამორეცხილი შიდა ზედაპირზე სულ მცირე 10 მლ სუფთა ტეტრაქლორეთილენით. შედეგად მიღებული მიქსტურა უნდა გაჩერდეს სულ მცირე 5 წუთი და შემდეგ თხევადი ნაწილი უნდა იქნეს მოშორებული ფრთხილი გაწურვით ისე, რომ არ მოხდეს ნალექის დაკარგვა; ნალექი უნდა შეგროვდეს გასაფილტრ ქაღალდზე, რომელიც მოთავსებულია ძაბრში, დარჩენილი TCE-ის გამოსაყოფად და ნალექში ცხიმის დალექვის თავიდან ასაცილებლად. ნალექი უნდა გაშრეს და დალექვის ეტაპის კონტროლისთვის რეკომენდებულია შემდეგ აიწონოს (0.001გ-იანი სიზუსტით). და ბოლოს ნალექი უნდა გაიცრას 0.25 მმ-ზე და გაისინჯოს შედეგად ორი მიღებული ფრაქცია, გარდა იმ შემთხვევისა, როდესაც გაცრა არ ჩაითვლება საჭიროდ; 2.1.3.3.5. მოტივტივე ნაწილაკების ექსტრაჰირება და მომზადება: ნალექის აღდგენის შემდეგ, ორი ფაზა უნდა დარჩეს სხვადასხვა ძაბრში: ტეტრაქლორეთილენის შემცველი სითხე და მყარი მასა მოტივტივე მასალით/ნივთიერებით. ეს მყარი ფაზა არის მოტივტივე ნაწილაკები (flotate) და იქნება აღდგენილი ძაბრიდან ტეტრაქლორეთილენის სრული გადაღვრით, ონკანის მოშვებით. ძაბრის გადატრიალებით, მოტივტივე ნაწილაკები გადატანილ იქნება დიდ პეტრის ფინჯანზე და ჰაერის დაშრობა მოხდება ორთქლის დეფლექტორის საშუალებით. თუ 5%-ზე მეტი მოტივტივე ნაწილაკები აღემატება 0.5მმ-ს, ის იცრება 0.25 მმ-ზე, მიღებული ორი ნაწილი კი უნდა შემოწმდეს; 2.1.3.3.6. ნედლი მასალის მომზადება – სულ მცირე 5 გ დაფქვილი ქვენიმუშის პორცია უნდა მომზადდეს. თუ მასალის 5%-ზე მეტი შეიცავს 0.50მმ-ზე მეტი ზომის ნაწილაკებს, ის იცრება 0.25მმ-ზე, მიღებული ორი ნაწილი კი უნდა შემოწმდეს. 2.1.3.4. ცხიმის ან ზეთის შემცველი ნიმუშების მომზადება ცხიმის ან ზეთის შემცველი ნიმუშების მომზადებისას დაცული უნდა იქნეს შემდეგი მოთხოვნები: ა) თუ ცხიმი არის მყარი, ის უნდა გალღვეს ღუმელში ვიდრე არ იქცევა სითხედ; ბ) პიპეტის გამოყენებით, ცხიმის ან ზეთის 40 მლ გადატანილი უნდა იქნეს ნიმუშის ძირიდან ცენტრიფუგირებისათვის გამოსაყენებელ ტუბში; გ) ცენტრიფუგირება 4000 ბრუნი/წუთში ხდება10 წუთის განმავლობაში; დ) ცენტრიფუგირების შედეგად თუ ქონი არის მყარი, ის უნდა გათბეს ღუმელში ვიდრე არ იქცევა სითხედ; ე) ცენტრიფუგირება მეორდება 5 წუთის განმავლობაში 4000 ბრუნი/წუთში; ვ) პატარა კოვზის ან შპადელის გამოყენებით, გამოწურული მინარევების ერთი მეორედი გადატანილი უნდა იქნეს მიკროსკოპულ სლაიდებზე შემოწმებისთვის, რეკომენდებულია გლიცეროლის გამოყენება, როგორც საფიქსაციო ნიადაგის (mounting medium); ზ) დარჩენილი მინარევები გამოყენებული უნდა იქნეს ნალექის მოსამზადებლად ამ თავის 2.1.3.3. პუნქტით დადგენილი მოთხოვნების შესაბამისად.
2.1.3.5. მღებავი რეაგენტების გამოყენება ცხოველური წარმოშობის შემადგენელი ნაწილაკების სწორი იდენტიფიცირებისათვის ოპერატორმა შესაძლოა გამოიყენოს მღებავი რეაგენტები ნიმუშის მომზადების დროს EURL-AP ის მიერ მომზადებული გაიდლაინების შესაბამისად. თუ ალიზარინის წითელი ხსნარი გამოყენებულია ნალექის შეღებვის მიზნით, დაცული უნდა იქნეს შემდეგი: ა) გამშრალი ნალექი გადატანილი უნდა იქნეს მინის სატესტო ტუბში და გასუფთავებულ იქნეს ორჯერ დაახლოებით 5მლ ეთანოლით (თითოეულ შემთხვევაში 30 წამიანი ნჯღრევით, გამხსნელი უნდა იქნეს გაჩერებული 1 წუთი და 30 წამი და შემდეგ გადაიღვაროს); ბ) ნალექი უნდა გათეთრდეს სულ მცირე 1 მლ ნატრიუმის ჰიპოქლორიდის ხსნარის დამატებით. რეაქცია უნდა გაგრძელდეს 10 წუთის განმავლობაში. ტუბი უნდა შეივსოს წყლით, ნალექი უნდა გაჩერდეს 2-3 წუთი და წყალი და ნაწილაკები ფრთხილად უნდა იქნეს გადმოღვრილი; გ) ნალექი უნდა გაირეცხოს კიდევ ორჯერ 10 მლ წყლით (სანჯღრეველა გამოყენებული იქნება 30 წამი, შემდეგ უნდა მოხდეს მისი გაჩერება და თითოეულ შემთხვევაში წყლის გადმოღვრა); დ) 2-დან 10 წვეთამდე ალიზარინის წითელი ხსნარი უნდა დაემატოს და მიქსტურა უნდა იქნეს მორეული. რეაქცია უნდა გაგრძელდეს 30 წამის განმავლობაში და შეფერილი ნალექი უნდა გაირეცხოს დაახლოებით ორჯერ 5 მლ ეთანოლით, რომელსაც უნდა მოჰყვეს გარეცხვა ერთხელ, აცეტონის მეშვეობით (თითოეულ შემთხვევაში 30 წამიანი ნჯღრევით, გამხსნელი უნდა გაჩერდეს დაახლოებით 1 წუთი და შემდეგ გადმოიღვაროს); ე) შეფერილი ნალექი უნდა გაშრეს.
2.1.4. მიკროსკოპიული გამოკვლევა 2.1.4.1. სლაიდის მომზადება მიკროსკოპიული სლაიდი უნდა მომზადდეს ნალექისგან და, ოპერატორის არჩევანის შესაბამისად ან მოტივტივე ნაწილაკებისაგან ან ნედლი მასალისგან. მომზადებული უნდა იქნეს სლაიდების საკმარისი რაოდენობა, რათა დარწმუნდეთ, რომ ამ დანართის 2.1.4.2. პუნქტით დადგენილი სრული შემოწმების პროტოკოლი შეიძლება იქნეს განხორციელებული; მიკროსკოპიული სლაიდები ფიქსირდება შესაბამის საფიქსაციო ნიადაგთან (mounting medium) ერთად EURL-AP ის მიერ დადგენილი SOP-ის შესაბამისად. სლაიდები იფარება მინასაფარით. 2.1.4.2. დაკვირვების დიაგრამა ცხოველის საკვების მასალასა და კომბინირებულ საკვებში ცხოველური წარმოშობის ნაწილაკების აღმოსაჩენად. მომზადებული მიკროსკოპული სლაიდებზე დაკვირვება უნდა განხორციელდეს ამ დანართის დიაგრამა №1-ში და დიაგრამა №2-ში მოცემული დაკვირვების დიაგრამების შესაბამისად. მიკროსკოპული დაკვირვებები უნდა განხორციელდეს მრავალფუნქციური მიკროსკოპის გამოყენებით ნალექზე და, ოპერატორის არჩევანის შესაბამისად, მოტივტივე ნაწილაკებზე ან ნედლ მასალაზე. დაუმუშავებელი ნაწილის მიმართ მრავალფუნქციურ მიკროსკოპთან ერთად შესაძლოა გამოყენებულ იქნეს სტერეომიკროსკოპი. თითოეული სლაიდი ექვემდებარება სრულ სკრინინგს სხვადასხვა სიდიდეზე (გადიდებაზე); ზუსტი განმარტებები იმის შესახებ, თუ როგორ გამოვიყენოთ დაკვირვების დიაგრამა, მოცემულია EURL-AP-ის მიერ შემუშავებულ SOP-ში, რომელიც გამოქვეყნებულია მის ვებგვერდზე. დასაკვირვებელი სლაიდების მინიმალური რიცხვი, პროტოკოლის თანახმად, დაკვირვების თითოეულ ეტაპზე უნდა იქნეს მკაცრად დაცული, გარდა იმ შემთხვევისა, როდესაც მასალის მთლიანი ფრაქცია არ იძლევა იმის შესაძლებლობას, რომ მოხდეს სლაიდების დადგენილი რაოდენობის მიღწევა, მაგალითად, როდესაც ნალექი საერთოდ არ მიიღება. არაუმეტეს 6 სლაიდი უნდა იქნეს დაკვირვებული თითოეულ კვლევაზე ნაწილაკების რაოდენობის ჩანაწერებისათვის. მოტივტივე ნაწილაკზე ან ნედლეულზე დამატებითი სლაიდების მომზადებისას გამოიყენება საფიქსაციო ნიადაგი (mounting medium) მღებავი მახასიათებლებით, როგორც ეს მოცემულია ამ დანართის 2.1.2.1.4. პუნქტში, სტრუქტურების (მაგ.: ბუმბულები, თმები, კუნთები ან სისხლის ნაწილაკები), რომლებიც გამოვლინდა სხვა საფიქსაციო ნიადაგით (mounting media) (როგორც ეს მოცემულია ამ დანართის 2.1.2.1.3. პუნქტში) მომზადებულ სლაიდებზე, შემდგომ დასახასიათებლად ნაწილაკების რაოდენობა დათვლილი უნდა იქნეს თითოეული სლაიდის რაოდენობის მიხედვით, არაუმეტეს 6 განსაზღვრისა, მათ შორის დამატებითი სლაიდები უფრო სპეციფიკური საფიქსაციო ნიადაგით (mounting medium). იმისათვის რომ ხელი შეეწყოს ნაწილაკების ბუნების იდენტიფიკაციას და წარმოშობას, ოპერატორმა შესაძლებელია გამოიყენოს ისეთი მხარდამჭერი მექანიზმები, როგორიცაა გამოსახულების ბიბლიოთეკები და რეფერენს (ეტალონური) ნიმუშები.
დიაგრამა №1
დაკვირვების დიაგრამა, რომელიც შეეხება პირველი განსაზღვრისას ცხოველის კომბინირებულ საკვებსა და ცხოველის საკვების მასალის გამოვლენას (D1 და D2 ეხება პირველ და მეორე განსაზღვრებებს; *: ხმელეთის ხერხემლიანები, თევზები)
დიაგრამა №2
დაკვირვების დიაგრამა, რომელიც შეეხება მეორე განსაზღვრისას ცხოველის კომბინირებული საკვებისა და ცხოველის საკვების მასალის გამოვლენას (D1 და D2 ეხება პირველ და მეორე განსაზღვრებებს; *: ხმელეთის ხერხემლიანი, თევზები)
2.1.4.3. გამოკვლევების რაოდენობა განსაზღვრა უნდა ჩატარდეს სხვადასხვა ქვენიმუშზე, რომელთა თითოეული წონა უნდა იყოს 50 გრ. თუ ამ დანართის დაკვირვების დიაგრამის №1-ის შესაბამისად განხორციელებული პირველი გამოკვლევის შემდეგ, ხმელეთის ცხოველის ან თევზის მახასიათებელი არცერთი ცხოველური ნაწილაკი არ აღმოჩნდა, არ არის საჭირო რაიმე დამატებითი კვლევა, გამოკვლევის შედეგი მოხსენებული იქნება ამ დანართის 2.1.5.1 პუნქტით დადგენილი ტერმინოლოგიით. თუ ამ დანართის დაკვირვების დიაგრამა №1-ის შესაბამისად განხორციელებული პირველი გამოკვლევის შემდეგ, გამოვლენილია მოცემული ბუნების (ხმელეთის ცხოველის ან თევზის) ერთი ან მეტი ცხოველის ნაწილაკი და ნაპოვნი ნაწილაკების ბუნება ადასტურებს ნიმუშის დეკლარირებულ შინაარსს, მეორე კვლევის ჩატარება აუცილებელი არ არის. თუ მოცემული ბუნების ცხოველების ნაწილაკების რაოდენობა, ამ პირველი გამოკვლევის დროს არის 5-ზე მეტი, გამოკვლევის შედეგი თითოეული ცხოველის ბუნებზე მოხსენებული იქნება ამ დანართის 2.1.5.3-ე პუნქტით დადგენილი ტერმინოლოგიით. სხვა შემთხვევაში, გამოკვლევის შედეგი თითოეულ ცხოველის ბუნებაზე, მოხსენებული უნდა იქნეს ამ დანართის 2.1.5.2-ე პუნქტით დადგენილი ტერმინოლოგიის შესაბამისად. სხვა შემთხვევებში, მათ შორის, როდესაც ლაბორატორიას არ მიეწოდება შემადგენლობის დეკლარაცია, უნდა განხორციელდეს მეორე კვლევა ახალი ქვენიმუშიდან. თუ ამ დანართის მე-2 დიაგრამაზე მოცემული დაკვირვების დიაგრამის შესაბამისად განხორციელებული მეორე გამოკვლევის შემდეგ, მოცემული ბუნების აღმოჩენილი ცხოველური ნაწილაკების ჯამი არის 10-ზე მეტი, გამოკვლევის შედეგი თითოეული ცხოველის ბუნებაზე მოხსენებული იქნება ამ დანართის 2.1.5.3. პუნქტით დადგენილი ტერმინოლოგიით. სხვა შემთხვევაში, გამოკვლევის შედეგი თითოეულ ცხოველის ბუნებაზე, მოხსენებული უნდა იქნეს ამ დანართის 2.1.5.2-ე პუნქტით დადგენილი ტერმინოლოგიის შესაბამისად.
2.1.5. შედეგების ფორმულირება შედეგების ფორმულირებისას (ჩანაწერების წარმოებისას), ლაბორატორია მიუთითებს რომელი ტიპის მასალაზე განხორციელდა გამოკვლევა (ნალექი, მოტივტივე ნაწილაკები ან ნედლი მასალა) და რამდენი კვლევა იქნა განხორციელებული. ანგარიშგებაში ნათლად უნდა იყოს მითითებული რამდენი განსაზღვრა იქნა ჩატარებული და ჩატარდა თუ არა ფრაქციების ფილტრაცია(გაცრა) სლაიდების მომზადებამდე, ამ დანართის 2.1.3.3.4-ე პუნქტის ბოლო ქვეპუნქტის შესაბამისად. ჩანაწერი უნდა მოიცავდეს სულ მცირე ინფორმაციას ხმელეთის ცხოველებისა და თევზის შემადგენელი ნაწილაკების არსებობის შესახებ. განსხვავებული შედეგები ფორმულირდება შემდეგნაირად: 2.1.5.1. აღნიშნული ბუნების არცერთი ცხოველური ნაწილაკი არ იქნა გამოვლენილი: – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში ხმელეთის ცხოველებიდან მიღებული არცერთი ნაწილაკი არ არის აღმოჩენილი; – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში თევზიდან მიღებული არცერთი ნაწილაკი არ არის აღმოჩენილი; 2.1.5.2. მოცემული ბუნების 1-დან 5 ცხოველის ნაწილაკი ვლინდება, როდესაც მხოლოდ ერთი განსაზღვრა ხორციელდება, ან მოცემული ბუნების 1-დან 10 ნაწილაკი ვლინდება, როდესაც ორი განსაზღვრა ხორციელდება (გამოვლენილი ნაწილაკების რაოდენობა მოცემულია გამოვლენის ზღვარს ქვემოთ, რომელიც განსაზღვრულია ევროკავშირის რეფერენს ლაბორატორიის სტანდარტულ ოპერაციების პროცედურებში (SOP) ცხოველის საკვების ცხოველური პროტეინებისთვის (EURL-AP) და რომელიც გამოქვეყნებულია მის ვებგვერდზე(24):
როდესაც შესრულებულია მხოლოდ ერთი განსაზღვრა: – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში, ხმელეთის ცხოველებიდან მიღებული 5 ნაწილაკზე მეტი არ აღმოჩენილა. ნაწილაკები იყო იდენტიფიცირებული როგორც ძვალი, ხრტილი, კუნთი, ბალანი, რქა და ა.შ. აღნიშნული დაბალი დონე, კერძოდ, მიკროსკოპიული მეთოდის გამოვლენის ზღვარს ქვემოთ ყოფნა მიუთითებს, რომ მცდარი პოზიტიური შედეგის რისკი არ შეიძლება იქნეს უგულებელყოფილი; – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში, თევზიდან მიღებული 5 ნაწილაკზე მეტი არ აღმოჩენილა. ნაწილაკები იყო იდენტიფიცირებული, როგორც თევზის ფხა, თევზის ქერცლი, ხრტილი, კუნთი, ოტოლიტები, ლაყუჩები და ა.შ. აღნიშნული დაბალი დონე, კერძოდ, მიკროსკოპიული მეთოდის გამოვლენის ზღვარს ქვემოთ ყოფნა მიუთითებს, რომ მცდარი პოზიტიური შედეგის რისკი არ შეიძლება იქნეს უგულებელყოფილი.
როდესაც შესრულებულია ორი განსაზღვრა: – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში, ორი განსაზღვრის შედეგად, ხმელეთის ცხოველებიდან მიღებული 10 ნაწილაკზე მეტი არ არის აღმოჩენილი; ნაწილაკები იყო იდენტიფიცირებული, როგორც ძვალი, ხრტილი, კუნთი, ბალანი, რქა და ა.შ.; აღნიშნული დაბალი დონე, კერძოდ, მიკროსკოპიული მეთოდის გამოვლენის ზღვარს ქვემოთ ყოფნა მიუთითებს, რომ მცდარი პოზიტიური შედეგის რისკი არ შეიძლება იქნეს უგულებელყოფილი. – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში, ორ განსაზღვარზე, თევზიდან მიღებული 10 ნაწილაკზე მეტუ არ აღმოჩენილა. ნაწილაკები იყო იდენტიფიცირებული, როგორც თევზის ფხა, თევზის ქერცლი, ხრტილი, კუნთი, ოტოლიტი ლაყუჩი და სხვ. აღნიშნული დაბალი დონე, კერძოდ, მიკროსკოპიული მეთოდის გამოვლენის ზღვარს ქვემოთ ყოფნა მიუთითებს, რომ მცდარი პოზიტიური შედეგის რისკი არ შეიძლება იქნეს უგულებელყოფილი.
დამატებით: – ნიმუშის წინასწარ გაცრის შემთხვევაში, ჩანაწერში აღნიშნული უნდა იქნეს თუ რომელ ფრაქციაში (ნაწილში) (გაცრილ, გრანულირებულ (პალეტირებული) ნაწილში თუ მარცვალში) იქნა აღმოჩენილი ცხოველური ნაწილაკები. ცხოველური ნაწილაკების აღმოჩენა მხოლოდ გაცრილ ნაწილში მიუთითებს გარემოს დაბინძურებაზე.
– როდესაც აღმოჩენილია მხოლოდ ცხოველური ნაწილაკები, რომელთა კატეგორიზაცია ვერ ხერხდება, როგორც ხმელეთის ცხოველები ან თევზები (მაგ., კუნთოვანი ბოჭკოები), ჩანაწერში უნდა აღინიშნოს, რომ ცხოველების მხოლოდ ასეთი ნაწილაკები იქნა აღმოჩენილი და არ შეიძლება მათი გამორიცხვა, რომ ისინი ხმელეთის ცხოველებიდან წარმოიშობა. 2.1.5.3. მოცემული ბუნების 5-ზე მეტი ცხოველის ნაწილაკი ვლინდება, როდესაც მხოლოდ ერთი განსაზღვრა ხორციელდება, ან მოცემული ბუნების 10-ზე მეტი ნაწილაკი ვლინდება ორი განსაზღვრის შემთხვევაში: როდესაც შესრულებულია მხოლოდ ერთი განსაზღვრა: – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში, ხმელეთის ცხოველებიდან მიღებული 5 ნაწილაკზე მეტი ნაწილაკი აღმოჩნდა. ნაწილაკები იყო იდენტიფიცირებული როგორც ძვალი, ხრტილი, კუნთი, ბალანი, რქა და ა.შ. – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში, თევზიდან მიღებული 5 ნაწილაკზე მეტია აღმოჩენილი. ნაწილაკები იყო იდენტიფიცირებული, როგორც თევზის ფხა, თევზის ქერცლი, ხრტილი, კუნთი, ოტოლიტები, ლაყუჩები და ა.შ.
როდესაც შესრულებულია ორი განსაზღვრა: – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში ორ განსაზღვრაზე ხმელეთის ცხოველებიდან მიღებული 5-ზე მეტი ნაწილაკია აღმოჩენილი. ნაწილაკები იყო იდენტიფიცირებული, როგორც ძვალი, ხრტილი, კუნთი, ბალანი, რქა და ა.შ.; – სინათლის მიკროსკოპის გამოყენებით, წარმოდგენილ ნიმუშში ორ განსაზღვრაზე თევზიდან მიღებული 10 ნაწილაკზე მეტია აღმოჩენილი. ნაწილაკები იყო იდენტიფიცირებული, როგორც თევზის ფხა, თევზის ქერცლი, ხრტილი, კუნთი, ოტოლიტი ლაყუჩი და სხვ.
დამატებით: – ნიმუშის წინასწარ გაცრის შემთხვევაში, ჩანაწერში აღნიშნული უნდა იქნეს თუ რომელ ნაწილში (გაცრილ, გრანულირებულ (პალეტირებული) ნაწილში თუ მარცვალში) იქნა აღმოჩენილი ცხოველური ნაწილაკები. ცხოველური ნაწილაკების აღმოჩენა მხოლოდ გაცრილ ნაწილში მიუთითებს გარემოს დაბინძურებაზე. – როდესაც აღმოჩენილია მხოლოდ ცხოველური ნაწილაკები, რომელთა კატეგორიზაცია ვერ ხერხდება, როგორც ხმელეთის ცხოველები ან თევზები (მაგ., კუნთოვანი ბოჭკოები), ჩანაწერში უნდა აღინიშნოს, რომ ცხოველების მხოლოდ ასეთი ნაწილაკები იქნა აღმოჩენილი და არ შეიძლება მათი გამორიცხვა, რომ ისინი ხმელეთის ცხოველებიდან წარმოიშობა.
2.2. PCR – პოლიმერაზული ჯაჭვური რეაქცია 2.2.1. გამოყენების პრინციპი ცხოველური წარმოშობის დეზოქსირიბონუკლეინის მჟავის (შემდგომში – DNA) ფრაგმენტები, რომელიც შესაძლოა წარმოდგენილი იყოს ცხოველის საკვებ მასალასა და კომბინირებულ საკვებში, შესაძლოა გამოვლინდეს სახეობა სპეციფიკური სამიზნე DNA-ის ფრაგმენტების გენეტიკური ამპლიფიკაციის ტექნიკით, PCR ის მეშვეობით.
PCR მეთოდი თავდაპირველად მოითხოვს DNA-ის ექსტრაციის ეტაპს, რასაც თან სდევს ექსტრაქციის შედეგად მიღებული დნმ-ის ამპლიფიკაციის ეტაპი სამიზნე დნმ-ით ცხოველის სახეობის დასადგენად. 2.2.2. რეაგენტები და აღჭურვილობა 2.2.2.1. რეაგენტები 2.2.2.1.1. რეაგენტები დნმ-ის მოპოვების ეტაპზე DNA-ის ექსტრაქციის ეტაპისათვის მხოლოდ ის რეაგენტები გამოიყენება, რომლებიც დადასტურებულია EURL-AP ის მიერ და გამოქვეყნებულია მის საიტზე; 2.2.2.1.2. რეაგენტები გენეტიკური გამაძლიერებელი ეტაპისთვის 2.2.2.1.2.1. პრაიმერები და ზონდები გენეტიკური ამპლიფიკაციის ეტაპისთვის მხოლოდ ის პრაიმერები და ზონდები გამოიყენება, რომლებსაც აქვთ ოლიგონუკლეოტიდის თანმიმდევრობა დადასტურებული EURL-AP ის მიერ (25); 2.2.2.1.2.2. მასტერ მიქსი მხოლოდ მასტერმიქსის ხსნარი, რომელიც არ შეიცავს რეაგენტებს, რომლებმაც შესაძლოა მცდარი შედეგი აჩვენონ ცხოველური DNA-ის არსებობის გამო26. 2.2.2.1.2.3. დეკონტამინაციის რეაგენტები 2.2.2.1.2.3.1. მარილმჟავას ხსნარი (0.1 N) 2.2.2.1.2.3.2. მათეთრებელი (ნატრიუმის ჰიპოქლორიდის ხსნარი 0.15% აქტიური ქლორზე) 2.2.2.1.2.3.3. არაკოროზიული რეაგენტები დეკონტამინაციის ძვირადღირებული მოწყობილობისთვის, ისეთი როგორიცაა ანალიზური სასწორები (მაგალითად, MP Biomedicals DNA Erase TM) 2.2.2.2. აღჭურვილობა 2.2.2.2.1. ანალიზური სასწორი 0.001 გრამის სიზუსტით; 2.2.2.2.2. საფქვავი მოწყობილობა; 2.2.2.2.3. თერმოციკლერი RT-PCR ის შესაძლებლობით; 2.2.2.2.4. მიკროცენტრიფუგა ცენტრიფუგას ტუბისათვის; 2.2.2.2.5. 1მლ-დან 1000 მლ-მდე მოცულობის მიკროპიპეტების ნაკრები; 2.2.2.2.6. პლასტმასის სტანდარტული მოლეკულურ ბიოლოგიური ჭურჭელი: მიკროცენტრიფუგის ტუბები, მიკროპიპეტის პლასტმასის ფილტრიანი წვერები, თერმოციკლერის თეფშები; 2.2.2.2.7. საყინულეები ნიმუშებისა და რეაგენტების შესანახად. 2.2.3. ნიმუშის აღება და მომზადება 2.2.3.1. ნიმუშის აღება 2.2.2.2. გამოკვლევისთვის გამოიყენებული უნდა იქნეს რეპრეზენტატიული ნიმუში. 2.2.3.2. ნიმუშის მომზადება DNA-ის ექსტრაქციამდე მზადდება ლაბორატორიული ნიმუში. სულ მცირე ნიმუშის 50 გრამი უნდა იქნეს გამოყენებული ქვენიმუშად ანალიზისა და შემდგომი დაფქვისთვის;
ნიმუშის მომზადება უნდა განხორციელდეს სათავსოში, სადაც DNA-ის ექსტრაქცია და ISO 24276 სტანდარტის შესაბამისად განხორციელებული გენეტიკური ამპლიფიკაციის რეაქციები არ ხორციელდება; უნდა მომზადდეს ორი სულ მცირე 100-100-მგ-იანი გამოსაკვლევი ნიმუშის პორცია.
2.2.4. DNA-ის ექსტრაქცია DNA-ის ექსტრაქცია უნდა განხორციელდეს EURL-AP-ის მიერ დადგენილი SOP-ის გამოყენებით მომზადებულ თითოეული გამოსაკვლევ ნიმუშის პორციაზე. თითოეული ექსტრაქციის სერიისთვის მომზადებული უნდა იქნეს ექსტრაქციის ორი კონტროლი ISO 24276 სტანდარტის შესაბამისად: – ექსტრაქციის კონტროლი; – დადებითი DNA-ის ექსტრაქციის კონტროლი.
2.2.5. გენეტიკური ამპლიფიკაცია გენეტიკური ამპლიფიკაცია უნდა განხორციელდეს იმ მეთოდების გამოყენებით, რომლებიც ვალიდირებულია იდენტიფიკაციის საჭიროების მქონე თითოეული სახეობისთვის. ეს მეთოდები განსაზღვრულია EURL-AP-ის მიერ დადგენილი SOP-ით. ინჰიბირების შეფასებისათვის თითოეული DNA-ის ექსტრაქცია უნდა გაანალიზდეს სულ მცირე ორ სხვადასხვა გაზავებულ ხსნარში.
ამპლიფიკაციის კონტროლი უნდა მომზადდეს ISO 24276 სტანდარტის შესაბამისად თითოეული სახეობისთის: – სამიზნე DNA-ის დადებითი კონტროლი გამოყენებული უნდა იქნეს PCR-ის თითოეული პლანშეტისთვის ან სერიისთვის; – ამპლიფიკაციის რეაგენტის კონტროლი გამოყენებული უნდა იქნეს PCR-ის კვლევის თითოეული პლანშეტისთვის ან სერიისთვის.
2.2.6. შედეგების ინტერპრეტაცია და ფორმულირება შედეგების რეპორტისას ლაბორატორიამ უნდა მიუთითოს სულ მცირე გამოყენებული გამოსაკვლევი ნიმუშის პორციის წონა და ექსტრაქციის ტექნიკა, ჩატარებული კვლევების რაოდენობა და დეტექციის მეთოდის ზღვარი.
შედეგები არ უნდა იქნეს ინტერპრეტირებული და ჩაწერილი თუ DNA-ის ექსტრაქციის და სამიზნე DNA-ის პოზიტიური კონტროლი არ უჩვენებს დადებით შედეგს სამიზნესთვის, მაშინ როცა ამპლიფიკაციის რეაგენტის კონტროლი ნეგატიურია.
თუ ორი გამოსაკვლევი პორციიდან შედეგები სხვადასხვაა (არ შეესაბამება ერთმანეთს), სულ მცირე გენეტიკური ამპლიფიკაციის ეტაპი უნდა განმეორდეს. თუ ლაბორატორია ეჭვობს, რომ შედეგების სხვადასხვაობის მიზეზი DNA-ის ექსტრაქტია, ახალი DNA-ის ექსტრაქცია და შემდგომი გენეტიკური ამპლიფიკაცია უნდა განხორციელდეს შედეგების ინტერპრეტაციამდე.
შედეგების საბოლოო ფორმულირება ეფუძნება ორი გამოსაკვლევი პორციის შედეგების ინტეგრაციასა და ინტერპრეტაციას EURL-AP ის მიერ დადგენილი SOP-ის შესაბამისად.
2.2.6.1. უარყოფითი შედეგი უარყოფითი შედეგი ანგარიშში მიეთითება შემდეგნაირად: წარმოდგენილ X ნიმუშიდან არ გამოვლინდა DNA (X – არის ცხოველთა სახეობა ან ცხოველთა სახეობის ჯგუფი, რომელზეც ტარდება გამოკვლევა).
2.2.6.2. დადებითი შედეგი დადებითი შედეგი ანგარიშში მიეთითება შემდეგნაირად: წარმოდგენილ X ნიმუშიდან გამოვლინდა DNA (X – არის ცხოველთა სახეობა ან ცხოველთა სახეობის ჯგუფი, რომელზეც ტარდება გამოკვლევა).
დანართი №7 ფრინველის საკვების ენერგეტიკული ღირებულების გამოანგარიშების მეთოდი 1. ენერგეტიკული ღირებულების გაანგარიშებისა და გამოხატვის მეთოდი ფრინველის კომბინირებული საკვების ენერგეტიკული ღირებულება უნდა გამოითვალოს ქვემოთ მოცემული ფორმულის შესაბამისად, საკვების ცალკეული ანალიტიკური კომპონენტების პროცენტული მაჩვენებლის საფუძველზე. აღნიშნული ღირებულება უნდა გამოიხატოს მეტაბოლიზირებული ენერგიის (ME) მეგაჯოულებში (MJ), აზოტზე შესწროებით, კომბინირებული საკვების ერთ კილოგრამზე:
ME-ს MJ/kg = 0.1551 ×% ნედლი ცილა + 0.3431 ×% ნედლი ცხიმი + 0.1669 ×% სახამებელი + 0.1301% შაქრის საერთო რაოდენობა (გამოხატულია საქაროზას სახით).
2. ტოლერანტობა, რომელიც გამოიყენება დეკლარირებულ ღირებულებებზე თუ სახელმწიფო კონტროლის შედეგად დადგინდება შეუსაბამობა (ცხოველის საკვების გაზრდილი ან შემცირებული ენერგეტიკული ღირებულება) შემოწმების შედეგსა და დეკლარირებულ ენერგეტიკულ ღირებულებას შორის, ნებადართული მინიმალური ტოლერანტობა იქნება 0,4 მგჯ/კგ ME.
3. შედეგების ფორმულირება ზემოაღნიშნული ფორმულის გამოყენების შემდეგ მიღებული შედეგი უნდა დამრგვალდეს მეათედებამდე.
4. ნიმუშის აღება და გამოკვლევის მეთოდები ცხოველის კომბინირებული საკვების ნიმუშის აღება და ანალიტიკური კომპონენტების შემცველობის განსაზღვრა, რომელიც მითითებულია გაანგარიშების მეთოდში, უნდა შესრულდეს ცხოველის საკვების სახელმწიფო კონტროლისთვის განსაზღვრული შესაბამისი ნიმუშის აღების მეთოდებისა გამოკვლევის მეთოდების მიხედვით.
გამოყენებული უნდა იქნეს შემდეგი:
– ნედლი ცხიმის შემცველობის დასადგენად: ნედლი ზეთებისა და ცხიმების განსაზღვრის მეთოდის B პრეცედურა, რომელიც მოცემულია ამ წესის დანართი №3-ის VIII თავში. – სახამებლის შემცველობის დასადგენად: პოლარიმეტრიული მეთოდი, რომელიც მოცემულია ამ წესის დანართი №3-ის XIIთავში.
დანართი №8
გამოკვლევის მეთოდები ცხოველის საკვებში უკვე ავტორიზაციის არმქონე უკანონოდ არსებული ცხოველის საკვები დანამატების კონტროლისთვის
თავი I მეთილის ბენზოქუატის განსაზღვრა
7-ბენზილოქსი-6-ბუტილ-3-მეტოქსიკარბონილ-4-ქინოლონი
1. მიზანი და გამოყენების ფარგლები ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ მეთილის ბენზოქუატის შემცველობა (ზღვარი) ცხოველის საკვებში. რაოდენობრივი შეფასების ლიმიტი შეადგენს 1 მგ/კგ.
2. გამოყენების პრინციპი მეთილ ბენზოქუატის ექსტრაჰირება ხდება ნიმუშიდან, რომელიც შეიცავს მეთანოლური მეთანსულფონის მჟავას ხსნარს. ექსტრაქტი იწმინდება დიქლორომეთანით, იონ-გაცვლითი ქრომატოგრაფიით და შემდეგ კვლავ დიქლორომეთანით. მეთილის ბენზოქუატის შემცველობა განისაზღვრება უკუქცევითი ფაზის მაღალი ხარისხის თხევადი ქრომატოგრაფიით (HPLC) UV დეტექტორით.
3. რეაგენტები 3.1. დიქლორომეთანი 3.2. მეთანოლი, HPLC ხარისხის ეკვივალენტი 3.3. HPLC მობილური ფაზა მეთანოლის (ამ თავის პუნქტი 3.2) და წყლის ნარევი (HPLC ხარისხის ეკვივალენტი) 75 + 25 (v + v). გაფილტრეთ 0,22 მკმ ფილტრის საშუალებით (ამ თავის პუნქტი 4.5) და მოახდინეთ ხსნარის დეგაზირება (მაგ., ულტრასონიფიკაციით 10 წუთის განმავლობაში). 3.4. მეთანსულფონის მჟავას ხსნარი, c = 2% 20,0 მლ მეთანსულფონის მჟავა განაზავეთ 1 000 მლ-მდე მეთანოლით (ამ თავის პუნქტი 3.2). 3.5. მარილმჟავას ხსნარი, c = 10% წყლით განაზავეთ 100 მლ მარილმჟავა (ρ20 1,18 გ/მლ) 1 000 მლ-მდე. 3.6. კათიონ-გაცვლითი ფისი Amberlite CG-120 (Na), 100-დან 200-მდე ხვრელით ფისის გამოყენებამდე ხდება მისი წინასწარი დამუშავება. 100 გ ფისოვანი ხსნარი გახსენით 500 მლ მარილმჟავას ხსნარით (ამ თავის პუნქტი 3.5) და გააცხელეთ ცხელ ფირფიტაზე ადუღებამდე ისე რომ თან ურიოთ შეუჩერებლად. მიეცით საშუალება გაცივდეს და მოახდინეთ მჟავას დეკანტირება (გამოცალკავება). ვაკუუმის ქვეშ გაფილტრეთ ფილტრის ქაღალდით. ფისი ორჯერ გარეცხეთ 500 მლ პორცია წყლით, შემდეგ კი 250 მლ მეთანოლით (ამ თავის პუნქტი 3.2). ჩამორეცხეთ ფისი დამატებით 250 მლ მეთანოლის პორციით და გააშრეთ გამფილტრავი მოწყობილობიდან (filter cake) გამომავალი ჰაერის ნაკადით. გამშარლი ფისი შეინახეთ თავსახურიან ბოთლში. 3.7. სტანდარტული ნივთიერება: სუფთა მეთილ ბენზოქატი (7-ბენზილოქსი-6-ბუტილ-3-მეთოქსიკარბონილ-4-ქინოლონი) 3.7.1. მეთილის ბენზოქუატის საწყისი სტანდარტული ხსნარი, 500 მკგ/მლ აწონეთ 0,1 მგ სიზუსტით 50 მგ სტანდარტული ნივთიერება (ამ თავის პუნქტი 3.7), გახსენით მეთანსულფონის მჟავას ხსნარში (ამ თავის პუნქტი 3.4) 100 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე და აურიეთ. 3.7.2. მეთილის ბენზოქუატის შუალედური სტანდარტული ხსნარი, 50 მკგ/მლ 5,0 მლ მეთილის ბენზოქუატის საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.7.1) გადაიტანეთ 50 მლ გრადირებულ კოლბაში, შეავსეთ მეთანოლით (ამ თავის პუნქტი 3.2) და აურიეთ. 3.7.3. საკალიბრო ხსნარები 1.0, 2.0, 3.0, 4.0 და 5.0 მლ მეთილის ბენზოქუატის შუალედური სტანდარტული ხსნარი (ამ თავის პუნქტი 3.7.2) გადაიტანეთ 25-მლ-იანი გრადირებული კოლბების სერიაში. შეავსეთ ნიშნულამდე მობილური ფაზით (ამ თავის პუნქტი 3.3) და აურიეთ. ამ ხსნარების კონცენტრაცია არის შესაბამისად, 2.0, 4.0, 6.0, 8.0 და 10.0 მკგ/მლ მეთილის ბენზოქუატი. აღნიშნული ხსნარები, გამოყენებამდე უნდა იყოს ახალი მომზადებული.
4. აპარატურა 4.1. ლაბორატორიული სადრვები 4.2. როტორული ამაორთქლებელი გადამღები მოწყობილობით 4.3. მინის სვეტი (250 მმ × 15 მმ), საცობით და დაახლოებით 200-მლ-იანი მოცულობის რეზერვუარით 4.4. HPLC მოწყობილობა ცვლადი ტალღის ულტრაიისფერი დეტექტორით ან დიოდური მატრიცული დეტექტორით 4.4.1. თხევადი ქრომატოგრაფიული სვეტი: 300 მმ × 4 მმ, C18, 10 მკმ შეფუთვა ან ეკვივალენტი 4.5. მემბრანული ფილტრები, 0.22 მკმ 4.6. მემბრანული ფილტრები, 0,45 მკმ
5. გამოყენების პროცედურა
5.1. ზოგადი პროცედურა 5.1.1. გამოკვლეული უნდა იქნეს ცხოველის საკონტროლო საკვები, მეთილის ბენზოქუატისა და ხელის შემშლელი ნივთიერებების არარსებობის შემოწმების მიზნით. 5.1.2. აღდგენითი ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების გამოკვლევის გზით, რომელიც გამდიდრებულია მეთილ ბენზოქუატის იმ რაოდენობის დამატებით, რომელიც ნიმუშში არსებულის ანალოგიურია. 15 მგ/კგ-მდე გამდიდრების მიზნით, 20 გ სუფთა ცხოველის საკვებს დაუმატეთ 600 მკლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.7.1), შეურიეთ და დაელოდეთ 10 წუთი, სანამ გააგრძელებდეთ ექსტრაჰირების ეტაპს (ამ თავის პუნქტი 5.2).
შენიშვნა: ამ მეთოდის მიზნებისათვის, ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის მსგავსი და გამოკვლევის შედეგად მეთილის ბენზოქუატი არ უნდა ვლინდებოდეს.
5.2. ექსტრაჰირება აწონეთ 0,01 გ სიზუსტით, დაახლოებით 20 გ მომზადებული ნიმუში და გადაიტანეთ 250 მლ კონუსურ კოლბაში. დაუმატეთ 100,0 მლ მეთანსულფონის მჟავას ხსნარი (ამ თავის პუნქტი 3.4) და შეანჯღრიეთ მექანიკურად (ამ თავის პუნქტი 4.1) 30 წუთის განმავლობაში. ხსნარი გაფილტრეთ ფილტრის ქაღალდის საშუალებით და ფილტრატი შეინახეთ სითხე-სითხის განცალკევების/დაყოფის (liquid-liquid partition step) ეტაპისთვის (ამ თავის პუნქტი 5.3).
5.3. სითხე-სითხის განცალკევების/დაყოფის ეტაპი (liquid-liquid partition step) 500-მლ-იან გამყოფ ძაბრში, რომელიც შეიცავს 100 მლ მარილმჟავას მჟავას ხსნარს (ამ თავის პუნქტი 3.5), გადაიტანეთ 25.0 მლ ფილტრატი, რომელიც მიღებული იქნა ამ თავის 5.2-ე პუნქტის მიხედვით. ძაბრში დაამატეთ 100 მლ დიქლორომეთანი (ამ თავის პუნქტი 3.1) და ანჯღრიეთ ერთი წუთის განმავლობაში. დააცადეთ, რომ ფენები განცალკავდეს და ქვედა ფენა (დიქლორომეთანი) გადავიდეს 500 მლ მრგვალძირიან კოლბაში. გაიმეორეთ წყლის ფაზის ექსტრაჰირება დიქლორომეთანის კიდევ 40 მლ პორციით და მოახდინეთ მათი შერწყმა პირველ ექსტრაქტთან მრგვალძირიან კოლბაში. აორთქლეთ დიქლორომეთანის ექსტრაქტი გამოშრობამდე როტორულ ამაორთქლებელზე (ამ თავის პუნქტი 4.2), რომელიც მუშაობს შემცირებული წნევის ქვეშ 40oC-ზე. გახსენით ნარჩენები 20-დან 25 მლ-მდე მეთანოლში (ამ თავის პუნქტი 3.2), კოლბას თავი დაუხურეთ და გააჩერეთ მთელი ექსტრაქტი იონ-გაცვლითი ქრომატოგრაფიისთვის (ამ თავის პუნქტი 5.4).
5.4. იონ-გაცვლითი ქრომატოგრაფია 5.4.1. კათიონ-გაცვლითი სვეტის მომზადება მინის სვეტის ქვედა ბოლოში მოათავსეთ მინაბოჭკოვანი ბამბა (ამ თავის პუნქტი 4.3). მოამზადეთ სუსპენზია 5,0 გ დამუშავებული კათიონ-გაცვლითი ფისით (ამ თავის პუნქტი 3.6) 50 მლ მარილმჟავათი (ამ თავის პუნქტი 3.5), ჩაასხით მინის სვეტში და დააცადეთ დალექვა. ფისოვანი ზედაპირის ოდნავ ზემოთა ნაწილიდან გადაწურეთ ზედმეტი მჟავა და გარეცხეთ სვეტი წყლით, სანამ ჩამონადენი არ შეიქნება ნეიტრალური ლაკმუსის ქაღალდთან მიმართებაში. 50 მლ მეთანოლი (ამ თავის პუნქტი 3.2) გადაიტანეთ სვეტზე და აცადეთ ჩამოწურვა ფისის ზედაპირზე.
5.4.2. სვეტის ქრომატოგრაფია მიღებული ექსტრაქტი პიპეტის საშუალებით ფრთხილად გადაიტანეთ სვეტზე (ამ თავის პუნქტი 5.3). მრგვალძირიანი კოლბა ჩამორეცხეთ 5-დან 10 მლ-მდე მოცულობის ორი პორცია მეთანოლით (ამ თავის პუნქტი3.2) და გადაიტანეთ ეს ჩამონარეცხები სვეტზე. დაიტანეთ ექსტრაქტი ფისის ზედაპირზე და ჩამორეცხეთ სვეტი 50 მლ მეთანოლით, დარწმუნდით, რომ დინების სიჩქარე არ აღემატებოდეს 5 მლ წუთში. მოაშორეთ ეფლუენტი. სვეტიდან მოახდინეთ მეთილის ბენზოქუატის ელუირება (გამორეცხვა) 150 მლ მეთანზულფონის მჟავის ხსნარის გამოყენებით (ამ თავის პუნქტი 3.4) და სვეტის ელუატის შეგროვება 250 მლ კონუსური ფორმის კოლბაში.
5.5. სითხე-სითხის განცალკევების/დაყოფის ეტაპი (liquid-liquid partition step) ელუატი, რომელიც მიღებული იქნა ამ თავის 5.4.2-ე პუნქტის მიხედვით, გადაიტანეთ 1 ლ-იან გამყოფ ძაბრში. ჩამორეცხეთ კონუსური კოლბა 5-დან 10 მლ-მდე მეთანოლით (ამ თავის პუნქტი 3.2) და ჩამონარეცხები შეურიეთ გამყოფი ძაბრის შემცველობასთან. დაამატეთ 300 მლ მარილმჟავას ხსნარი (ამ თავის პუნქტი 3.5) და 130 მლ დიქლორომეთანი (ამ თავის პუნქტი 3.1). ანჯღრიეთ 1 წუთის განმავლობაში და დააცადეთ, რომ ფენები განცალკავდეს და რომ ქვედა ფენა (დიქლორომეთანი) გადავიდეს 500 მლ მრგვალძირიან კოლბაში. გაიმეორეთ წყლის ფაზის ექსტრაჰირება დიქლორომეთანის კიდევ 70 მლ პორციით და მოახდინეთ მათი შერწყმა პირველ ექსტრაქტთან მრგვალძირიან კოლბაში. აორთქლეთ დიქლორომეთანის ექსტრაქტი გამოშრობამდე როტორულ ამაორთქლებელზე (ამ პუნქტის პუნქტი 4.2), რომელიც მუშაობს შემცირებული წნევის ქვეშ 40oC-ზე. გახსენით ნაშთი (ნარჩენები) დაახლოებით 5 მლ-მდე მეთანოლში (ამ თავის პუნქტი 3.2) და რაოდენობრივად გადაიტანეთ ეს ხსნარი 10 მლ გრადირებულ კოლბაში. ჩამორეცხეთ მრგვალძირიანი კოლბა დამატებით 1-დან 2 მლ-მდე მეთანოლის ორი პორციით და გადაიტანეთ გრადირებულ კოლბაში. შეავსეთ ნიშნულამდე მეთანოლით და აურიეთ. გაფილტრეთ ალიქვოტის პორცია მემბრანული ფილტრის (ამ თავის პუნქტი 4.6) მეშვეობით. შეინახეთ ეს ხსნარი HPLC-ის განსაზღვრისთვის (ამ თავის პუნქტი 5.6).
5.6. HPLC-ის განსაზღვრა
5.6.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან. – თხევადი ქრომატოგრაფიული სვეტი (ამ თავის პუნქტი 4.4.1) – HPLC მობილური ფაზა: მეთანოლის წყლის ნარევი (ამ თავის პუნქტი 3.3), – ნაკადის სიჩქარე: 1-დან 1,5 მლ/წუთში, – გამოვლენის ტალღის სიგრძე: 265 ნმ, – ინჯექტირების მოცულობა: 20-დან 50 მკლ-მდე.
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, რამდენჯერმე მოახდინეთ 4 მკგ/მლ შემცველობის მქონე საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.7.3), სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები და შეკავების სტაბილური დრო.
5.6.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.7.3) და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი,საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბცისასი.
5.6.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის ინჯექტირება (ამ თავის პუნქტი 5.5), იმავე მოცულობის გამოყენებით, რაც აღებული იქნა საკალიბრო ხსნარებისთვის და განსაზღვრეთ მეთილის ბენზოქუატის მწვერვალების საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების ფორმულირება განსაზღვრეთ ნიმუშის ხსნარის კონცენტრაცია მკგ/მლ-ში ნიმუშის ხსნარის მეთილის ბენზოქუატის პიკების საშუალო სიმაღლიდან (ფართობიდან) საკალიბრო მრუდის (ამ თავის პუნქტი 5.6.2) მითითებით.
მეთილის ბენზოქუატის ნიმუშის შემცველობა w (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: c = მეთილის ბენზოქუატის კონცენტრაციის ნიმუშის ხსნარი მკგ/მლ-ში, m = გამოსაკვლევი პორციის წონა გრამებში.
7. შედეგების ვალიდაცია 7.1. იდენტიფიკაცია ანალიტის იდენტურობა შესაძლებელია დადასტურდეს თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც ხდება ნიმუშის ექსტრაქტის სპექტრებისა და 10 მკგ/მლ-ის შემცველობის საკალიბრო ხსნარის (ამ თამის პუნქტი 3.7.3) შედარება.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი გამდიდრებულია შესაბამისი რაოდენობის შუალედური სტანდარტული ხსნარის (ამ თავის პუნქტი 3.7.2) დამატებით. მეთილის ბენზოქუატის დამატებული რაოდენობა უნდა იყოს მეთილის ბენზოქუატის სავარაუდო (შეფასებული) რაოდენობის იდენტური, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ მეთილის ბენზოქუატის პიკის სიმაღლე. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს თავდაპირველი სიგანის 10%-ის ფარგლებში.
7.1.2. გამოვლენა დიოდური – მატრიცის გამოყენებით შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით:
ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბსორბაციის (შეწოვის) ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, იდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცის გამოვლენისთვის ეს ჩვეულებრივ 2 ნმ-ის ფარგლებშია;
ბ) 220-დან 350 ნმ-მდე, ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100%-მდე ფარდობითი აბსორბაციის (შეწოვის) დიაპაზონში. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული ანალიტის შთანთქმის 15%-ს; გ) 220-დან 350 ნმ-მდე, ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და, როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15%-ს.
თუ რომელიმე ამ კრიტერიუმებიდან არ სრულდება, ანალიტის არსებობა არ დასტურდება.
7.2. განმეორებადობა ერთი და იმავე ნიმუშზე ჩატარებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: 4მგ/კგ-სა და 20 მგ/კგ შუალედში მეთილის ბენზოქუატის შემცველობის უფრო მაღალი შედეგის 10%-ს.
7.3. აღდგენა გამდიდრებული საკონტროლო ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 90%.
8. ერთობლივი კვლევის შედეგები 10 ლაბორატორიამ გამოიკვლია ხუთი ნიმუში. თითოეულ ნიმუშზე ჩატარდა განმეორებითი (დუბლიკატი) გამოკვლევა.
ND = არ არის გამოვლენილი sr = განმეორებადობის სტანდარტული გადახრა, CVr = განმეორებადობის ვარიაციის კოეფიციენტი, % SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი %.
თავი II ოლაქინდოქსის განსაზღვრა
2 – [N-2 ' – (ჰიდროქსიეთილ) კარბამოილი] -3-მეთილქინოქსალინი-N1, N4 -დიოქსიდი
1. მიზანი და გამოყენების ფარგლები ეს მეთოდი საშუალებას იძლევა მოვახდინოთ ცხოველის საკვებში ოლაქინდოქსის შემცველობის განსაზღვრა. რაოდენობრივი შეფასების ზღვარი შეადგენს 5 მგ/კგ-ს.
2. გამოყენების პრინციპი ნიმუში ექსტრაჰირება ხდება წყალ-მეთანოლის ნარევით. ოლაქინდოქსის შემცველობა განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (HPLC), UV-ის (ულტრაიისფერის) საშუალებით.
3. რეაგენტები 3.1. მეთანოლი. 3.2. მეთანოლი, რომელიც ეკვივალენტურია HPLC ხარისხის. 3.3. წყალი, რომელიც ეკვივალენტურია HPLC ხარისხის. 3.4. მობილური ფაზა HPLC-სთვის. წყალი (ამ თავის პუნქტი 3.3) – მეთანოლის (ამ თავის პუნქტი 3.2) ნარევი, 900 +100 (V + V). 3.5. სტანდარტული ნივთიერება: სუფთა ოლაქინდოქსი 2-[N-2’-(ჰიდროქსიეთილ) კარბამოილი] – 3-მეთილქინოქსალინი-N1, N4 -დიოქსიდი, E 851. 3.5.1. ოლაქინდოქსის საწყისი სტანდარტული ხსნარი, 250 მკგ/მლ აწონეთ 0,1 სიზუსტით მგ 50 მგ ოლაქინდოქსი (ამ თავის პუნქტი 3.5) 200 მლ გრადირებულ კოლბაში და დაამატეთ დაახლოებით 190 მლ წყალი. შემდეგ მოათავსეთ კოლბა 20 წთ. ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.1). ულტრაბგერითი დამუშავების შემდეგ ხსნარი მიიყვანეთ ოთახის ტემპერატურაზე, შეავსეთ ნიშნულამდე წყლით და აურიეთ. შეახვიეთ კოლბა ალუმინის ფოლგით და შეინახეთ მაცივარში. აღნიშნული ხსნარები, ყოველ თვე უნდა იყოს ახალი მომზადებული. 3.5.2. ოლაქინდოქსის შუალედური სტანდარტული ხსნარი, 25 მკგ/მლ 10,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.5.1) გადაიტანეთ 100 მლ გრადირებულ კოლბაში, მობილური ფაზით შეავსეთ ნიშნულამდე (ამ თავის პუნქტი 3.4) და აურიეთ. შეახვიეთ კოლბა ალუმინის ფოლგით და შეინახეთ მაცივარში. ეს ხსნარი ყოველდღე ახალი უნდა მომზადდეს. 3.5.3. საკალიბრო ხსნარები 50-მლ-იანი გრადირებული კოლბების სერიაში გადაიტანეთ 1,0, 2,0, 5,0, 10,0, 15,0 და 20,0 მლ შუალედური სტანდარტული ხსნარი (ამ თავის პუნქტი 3.5.2). მობილური ფაზით შეავსეთ ნიშნულამდე (ამ თავის პუნქტი 3.4) და აურიეთ. შეახვიეთ კოლბები ალუმინის ფოლგით. ეს ხსნარები შეესაბამება 0,5, 1,0, 2,5, 5,0, 7,5 და 10,0 მკგ ოლაქინდოქსს მლ შესაბამისად. ეს ხსნარები ყოველდღე ახალი უნდა მომზადდეს.
4. აპარატურა 4.1. ულტრაბგერითი აბაზანა 4.2. მექანიკური სადღვები 4.3. HPLC მოწყობილობა ცვალებადი ტალღის სიგრძის ულტრაიისფერი დეტექტორით ან დიოდური მატრიცის დეტექტორით 4.3.1. თხევადი ქრომატოგრაფიული სვეტი, 250 მმ × 4 მმ, C18, 10 მკმ შეფუთვა, ან ეკვივალენტი 4.4. მემბრანული ფილტრები, 0,45 მკმ.
5. პროცედურა შენიშვნა: ოლაქინდოქსი მგრძნობიარეა სინათლის მიმართ. ჩაატარეთ ყველა პროცედურა სუსტი შუქის ქვეშ ან გამოიყენეთ ქარვის მინის ჭურჭელი.
5.1. ზოგადი პროცედურა 5.1.1. გამოკვლეული უნდა იქნეს ცხოველის საკონტროლო საკვები, რათა შემოწმდეს ოლაქინდოქსისა და ხელის შემშლელი ნივთიერებების არსებობა.
5.1.2. აღდგენითი ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების (ამ თავის პუნქტი 5.1.1) გამოკვლევით, რომელიც გამდიდრებულია ოლაქინდოქსის რაოდენობის დამატებით, რაც ნიმუშში არსებულის იდენტურია. 50 მგ/კგ დონეზე გამდიდრების მიზნით, 10,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.5.1) გადაიტანეთ 250 მლ კონუსური ფორმის კოლბაში. აორთქლეთ გამხსნელი დაახლოებით 0,5 მლ-მდე. დაუმატეთ 50 გრ ცხოველის საკონტროლო საკვები, აურიეთ და დაელოდეთ 10 წუთს, სანამ გადახვალთ ექსტრაჰირების (ამ თავის პუნქტი 5.2) ეტაპზე. შენიშვნა: ამ მეთოდის მიზნებისათვის ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის მსგავსი და ოლაქინდოქსის კვლევისას არ უნდა ვლინდებოდეს.
5.2. ექსტრაჰირება აწონეთ 0,01 გ-მდე სიზუსტით, დაახლოებით 50 გ ნიმუში. გადაიტანეთ 1 000 მლ კონუსურ კოლბაში და დაუმატეთ 100,0 მლ მეთანოლი (ამ თავის პუნქტი 3.1) და მოათავსეთ კოლბა 5 წუთით ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.1). დაამატეთ 410 მლ წყალი და დატოვეთ ულტრაბგერით აბაზანაში კიდევ 15 წთ. ამოიღეთ კოლბა ულტრაბგერითი აბაზანიდან, შეანჯღრიეთ იგი 30 წუთის განმავლობაში სადღვებზე (ამ თავის პუნქტი 4.2) და გაფილტრეთ დაკეცილი ფილტრის საშუალებით. 10,0 მლ ფილტრატი გადაიტანეთ 20-მლ-იან გრადირებულ კოლბაში, ნიშნულამდე შეავსეთ წყლით და აურიეთ. ალიქვოტი გაფილტრეთ მემბრანული ფილტრის (ამ თავის პუნქტი 4.4) საშუალებით. (იხ. ამ თავის პუნქტი 9. დაკვირვება) გადადით HPLC განსაზღვრებაზე (ამ თავის პუნქტი 5.3).
5.3. HPLC განსაზღვრა 5.3.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან.
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, საკალიბრო ხსნარის რამდენჯერმე (ამ თავის პუნქტი 3.5.3) შეყვანით, რომელიც შეიცავს 2,5 მკგ/მლ-ს, სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები და შენარჩუნების დრო.
5.3.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.5.3) და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი, საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბცისასი.
5.3.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის ინჯექტირება (ამ თავის პუნქტი 5.2), იმავე მოცულობის გამოყენებით, რაც აღებული იქნა საკალიბრო ხსნარებისთვის და განსაზღვრეთ ოლაქინდოქსის სიმაღლეების საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების ფორმულირება განსაზღვრეთ ნიმუშის ხსნარის კონცენტრაცია მკგ/მლ-ში, ნიმუშის ხსნარის ოლაქინდოქსის სიმაღლეების საშუალო სიმაღლიდან (ფართობიდან) საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.3.2). ოლაქინდოქსის ნიმუშის შემცველობა w (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ხსნარის ოლაქინდოქსის კონცენტრაცია (ამ თავის პუნქტი 5.2) მკგ/მლ-ში, m = გამოსაკვლევი პორციის წონა გრამებში (ამ თავის პუნქტი 5.2).
7. შედეგების ვალიდაცია
7.1. იდენტიფიკაცია ანალიტის იდენტურობა შეიძლება დადასტურდეს თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც ხდება ნიმუშის ექსტრაქტის (ამ თავის პუნქტი 5.2) სპექტრებისა და 5 მკგ/მლ-ის შემცველობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.5.3) შედარება.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი (ამ თავის პუნქტი 5.2) გამდიდრებულია შესაბამისი რაოდენობის საკალიბრო ხსნარის დამატებით (ამ თავის პუნქტი 3.5.3). ოლაქინდოქსის დამატებული რაოდენობა უნდა იყოს ოლაქინდოქსის რაოდენობის მსგავსი, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ ოლაქინდოქსის პიკის სიმაღლე. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს ოლაქინდოქსის მწვერვალის თავდაპირველი სიგანის არაგამდიდრებული ნიმუშის ექსტრაქტის ± 10%-ში ფარგლებში.
7.1.2. გამოვლენა დიოდური – მატრიცის გამოყენებით შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით: ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბსორბაციის (შეწოვის) ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, იდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცის გამოვლენისთვის ეს ჩვეულებრივ ±2 ნმ-ის ფარგლებშია; ბ) 220-დან 400 ნმ-მდე, ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100% ფარდობითი აბსორბაციის (შეწოვის) დიაპაზონში. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული ანალიტის შთანთქმის 15%-ს; გ) 220-დან 400 ნმ-მდე, ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15% -ს.
თუ ამ კრიტერიუმებიდან რომელიმე არ სრულდება, ანალიტის არსებობა არ დასტურდება.
7.2. განმეორებადობა ერთი და იმავე ნიმუშზე ჩატარებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა უფრო მაღალ შედეგთან შედარებისას არ უნდა აღემატებოდეს 15%-ს ოლაქინდოქსის 10-დან 200 მგ/კგ-მდე შემცველობისას.
7.3. აღდგენა გამდიდრებული საკონტროლო ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 90%.
8. ერთობლივი კვლევის შედეგები მოეწყო EC-ს ერთობლივი კვლევა 13-მდე ლაბორატორიის მიერ, რომელშიც გამოკვლეული იქნა გოჭის ოთხი ცხოველის საკვების ნიმუში, მათ შორის ერთი საკონტროლო ცხოველის საკვები. შედეგები მოცემულია ქვემოთ:
L = ლაბორატორიების რაოდენობა n = ერთეულების მნიშვნელობების რაოდენობა s r = განმეორებადობის სტანდარტული გადახრა S R = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა CVr = განმეორებადობის ვარიაციის კოეფიციენტი CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი.
9. დაკვირვება მიუხედავად იმისა, რომ მეთოდი ვალიდირებული არ არის ცხოველის საკვებში, რომელიც შეიცავს 100 მგ/კგ-ზე მეტ ოლაქინდოქსს, შესაძლებელია დამაკმაყოფილებელი შედეგების მიღება მცირე ზომის ნიმუშის აღებით ან/და ექსტრაქტის (ამ თავის პუნქტი 5.2) განზავებით საკალიბრო მრუდის (ამ თავის პუნქტი 5.3.2) დიაპაზონში კონცენტრაციის მისაღწევად.
თავი III ამპროლიუმის განსაზღვრა
1 – [(4-ამინო-2-პროპილპირიმიდინ-5-ილ) მეთილ] -2-მეთილ-პირიდინიუმის ქლორიდის ჰიდროქლორიდი
1. მიზანი და გამოყენების სფერო ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ ამპროლიუმის შემცველობა ცხოველის საკვებსა და პრემიქსებში. გამოვლენის ზაღვარი შეადგენს 1 მგ/კგ-ს, ხოლო რაოდნობრივი ზღვარი 5 მგ/კგ.
2. გამოყენების პრინციპი ნიმუშიის ექსტრაჰირება ხდება მეთანოლ-წყლის ნარევით. მობილური ფაზისა და მემბრანული ფილტრაციით განზავების შემდეგ, ამპროლიუმის შემცველობა განისაზღვრება კათიონის გაცვლის მაღალი ხარისხის თხევადი ქრომატოგრაფიით (HPLC) UV დეტექტორის გამოყენებით.
3. რეაგენტები 3.1. მეთანოლი. 3.2. აცეტონიტრილი, HPLC ხარისხის ეკვივალენტური. 3.3. წყალი, HPLC ხარისხის ეკვივალენტური. 3.4. ნატრიუმის დიჰიდროფოსფატის ხსნარი, c = 0,1 მოლი/ლიტრი. 1-მლ-იან გრადირებულ კოლბაში, წყალში (ამ თავის პუნქტი 3.3) გახსენით 13,80 გ ნატრიუმის დიჰიდროფოსფატის მონოჰიდრატი, შეავსეთ ნიშნულამდე წყლით (ამ თავის პუნქტი 3.3) და აურიეთ. 3.5. ნატრიუმის პერქლორატის ხსნარი, c = 1,6 მოლი/ლიტრი. 1 000 მლ გრადირებულ კოლბაში, წყალში (ამ თავის პუნქტი 3.3) გახსენით 224,74 გ ნატრიუმის პერქლორატის მონოჰიდრატი, შეავსეთ ნიშნულამდე წყლით (ამ თავის პუნქტი 3.3) და აურიეთ. 3.6. მობილური ფაზა HPLC-თვის (იხ. ამ თავის პუნქტი 9.1 დაკვირვება). აურიეთ აცეტონიტრილი (ამ თავის პუნქტი 3.2), ნატრიუმის დიჰიდროფოსფატის ხსნარი (ამ თავის პუნქტი 3.4) და ნატრიუმის პერქლორატის ხსნარი (ამ თავის პუნქტი 3.5), 450 + 450 + 100 (v + v + v). გამოყენებამდე გაფილტრეთ 0,22 მკმ მემბრანული ფილტრის (ამ თავის პუნქტი 4.3) საშუალებით და მოახდინეთ ხსნარის დეგაზირება (მაგ., ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.4) არანაკლებ 15 წუთის განმავლობაში). 3.7. სტანდარტული ნივთიერება: სუფთა ამპროლიუმი, 1 – [(4-ამინო-2-პროპილპირიმიდინი-5-ილ) მეთილ] -2-მეთიპლპირიდინიუმ ქლორიდი ჰიდროქლორიდი, E 750 (იხ. ამ თავის პუნქტი 9.2). 3.7.1. ამპროლიუმის საწყისი სტანდარტული ხსნარი, 500 მკგ/მლ აწონეთ 0,1 მგ სიზუსტით, 50 მგ ამპროლიუმი (ამ თავის პუნქტი 3.7) 100 მლ გრადირებულ კოლბაში, გახსენით 80 მლ მეთანოლში (ამ თავის პუნქტი 3.1) და განათავსეთ კოლბა 10 წთ ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.4). ულტრაბგერითი დამუშავების შემდეგ ხსნარი მიიყვანეთ ოთახის ტემპერატურამდე, შეავსეთ ნიშნულამდე წყლით და აურიეთ. ხსნარი სტაბილურია ≤ 4oC ტემპერატურაზე 1 თვის განმავლობაში. 3.7.2. ამპროლიუმის შუალედური სტანდარტული ხსნარი, 50 მკგ/მლ პიპეტით გადაიტანეთ 5,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.7.1) 50 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე ექსტრაჰირებული გამხსნელით (ამ თავის პუნქტი 3.8) და აურიეთ. ხსნარი სტაბილურია ≤ 4oC ტემპერატურაზე 1 თვის განმავლობაში.
3.7.3. საკალიბრო ხსნარები 0,5, 1,0 და 2,0 მლ შუალედური სტანდარტული ხსნარი (ამ თავის პუნქტი 3.7.2) გადაიტანეთ 50 მლ სერიის გრადირებულ კოლბებში. შეავსეთ ნიშნულამდე მობილური ფაზით (ამ თავის პუნქტი 3.6) და აურიეთ. შესაბამისად, ეს ხსნარები შეესაბამება 0,5, 1,0 და 2,0 მკგ ამპროლიუმს მლ-ზე. აღნიშნული ხსნარები, გამოყენებამდე უნდა იყოს ახალი მომზადებული. 3.8. ექსტრაქციის გამხსნელი მეთანოლი (ამ თავის პუნქტი 3.1) – წყლის ნარევი 2 + 1 (v + v).
4. აპარატურა 4.1. ინჯექტირების სისტემის მქონე HPLC მოწყობილობა, შესაფერისია 100 მკლ ინჯექტირების მოცულობებისთვის. 4.1.1. თხევადი ქრომატოგრაფიული სვეტი 125 მმ × 4 მმ, კათიონის გაცვლა Nucleosil 10 SA, 5 ან 10 მკმ შეფუთვა, ან ეკვივალენტი. 4.1.2. UV დეტექტორი ტალღის ცვალებადი რეგულირების ან დიოდური მატრიცის დეტექტორით. 4.2. მემბრანული ფილტრი, PTFE მასალა, 0,45 მკმ. 4.3. მემბრანული ფილტრი, 0,22 მკმ. 4.4. ულტრაბგერითი აბაზანა. 4.5. მექანიკური სადღვები ან მაგნიტური სათქვეფელა.
5. პროცედურა 5.1. ზოგადი პრინციპი 5.1.1. ცხოველის საკონტროლო საკვები აღდგენითი ტესტის (ამ თავის პუნქტი 5.1.2) შესასრულებლად უნდა მოხდეს ცხოველის საკონტროლო საკვების გამოკვლევა, რათა შემოწმდეს ამპროლიუმისა და ხელის შემშლელი ნივთიერებების არარსებობა. ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის მსგავსი და ამპროლიუმი ან ხელის შემშლელი ნივთიერებები არ უნდა გამოვლინდეს.
5.1.2. აღდგენითი ტესტი აღდგენითი ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების გამოკვლევით, რომელიც გამდიდრებულია ამპროლიუმის რაოდენობის დამატებით, რომელიც ნიმუშში არსებულის ანალოგიურია. 100 მგ/კგ დონემდე გამდიდრებისთვის, გადაიტანეთ 10.0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.7.1) 250 მლ კონუსური ფორმის კოლბაში და აორთქლეთ ხსნარი დაახლოებით 0,5 მლ-მდე. დაამატეთ 50 გ ცხოველის საკვები, კარგად აურიეთ და გააჩერეთ 10 წთ, ამავდროულად, ვიდრე გადახვიდოდეთ ექსტრაქციის ეტაპზე (ამ თავის პუნქტი 5.2), კვლავ აურიეთ რამდენჯერმე.
ალტერნატიულად, თუ ნიმუშის მსგავსი ტიპის ცხოველის საკონტროლო საკვები არ არის ხელმისაწვდომი (იხ. ამ თავის პუნქტი 5.1.1), აღდგენითი ტესტი შეიძლება ჩატარდეს სტანდარტული დამატების მეთოდით. ამ შემთხვევაში, ხდება გამოსაკვლევი ნიმუშის გამდიდრება აპროლიუმის რაოდენობით, ისე, როგორც ეს უკვე წარმოდგენილია ნიმუშში. ხდება ამ ნიმუშის გამოკვლევა გაუმდიდრებელ ნიმუშთან ერთად და აღდგენის გამოანგარიშება შესაძლებელია გამოკლებით.
5.2. ექსტრაჰირება 5.2.1. პრემიქსები (შემცველობა <1% ამპროლიუმი) და ცხოველის საკვები აწონეთ 0,01 გ სიზუსტით, 5-40 გ ნიმუში, ამპროლიუმის შემცველობიდან გამომდინარე, 500 მლ კონუსური ფორმის კოლბაში და დაამატეთ 200 მლ ექსტრაქციის გამხსნელი (ამ თავის პუნქტი 3.8). კოლბა მოათავსეთ ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.4) და გააჩერეთ 15 წუთის განმავლობაში. ამოიღეთ კოლბა ულტრაბგერითი აბაზანიდან და ანჯღრიეთ იგი 1 საათის განმავლობაში სადღვებზე ან ურიეთ მაგნიტური სათქვეფელათი (ამ თავის პუნქტი 4.5). განაზავეთ ექსტრაქტის ალიქვოტი მობილური ფაზით (ამ თავის პუნქტი 3.6) 0,5-2 მკგ/მლ ამპროლიუმის შემცველობასთან და აურიეთ (იხ. ამ თავის პუნქტი 9.3 დაკვირვება). არნიშნული 5-10 მლ განზავებული ხსნარი გაფილტრეთ მემბრანულ ფილტრზე (ამ თავის პუნქტი 4.2). გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.3).
5.2.2. პრემიქსები (ამპროლიუმი ≥ 1% შემცველობა) აწონეთ 0.001 გ სიზუსტით, 1-4 გ პრემიქსი, ამპროლიუმის შემცველობიდან გამომდინარე, 500 მლ კონუსური ფორმის კოლბაში და დაამატეთ 200 მლ ექსტრაქციის გამხსნელი (ამ თავის პუნქტი 3.8). კოლბა მოათავსეთ ულტრაბგერით აბაზანაში (ამ თავის პუნქტი 4.4) და გააჩერეთ 15 წუთის განმავლობაში. ამოიღეთ კოლბა ულტრაბგერითი აბაზანიდან და ანჯღრიეთ იგი 1 საათის განმავლობაში სადღვებზე ან ურიეთ მაგნიტურ სადღვებელაზე (ამ თავის პუნქტი 4.5). განაზავეთ ექსტრაქტის ალიქვოტი მობილური ფაზით (ამ თავის პუნქტი 3.6) 0,5-2 მკგ/მლ ამპროლიუმის შემცველობასთან და აურიეთ. ეს 5-10 მლ განზავებული ხსნარი გაფილტრეთ მემბრანულ ფილტრზე (ამ თავის პუნქტი 4.2). გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.3).
5.3. HPLC განსაზღვრა 5.3.1. პარამეტრები: სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები. შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი მოგვცემენ ეკვივალენტურ შედეგებს.
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, რამდენჯერმე მოახდინეთ საკალიბრო ხსნარი შეყვანა (ინჯექტირება) (ამ თავის პუნქტი 3.7.3), რომელიც შეიცავს 1,0 მკგ/მლ, სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები და შენარჩუნების დრო.
5.3.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.7.3) და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი,საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბცისასი.
5.3.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის ინჯექტირება (ამ თავის პუნქტი 5.2), იმავე მოცულობის გამოყენებით, რაც აღებული იქნა საკალიბრო ხსნარებისთვის და განსაზღვრეთ ამპროლიუმის მწვერვალების საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების გაანგარიშება განსაზღვრეთ ნიმუშის ხსნარის კონცენტრაცია მკგ/მლ-ში, ნიმუშის ხსნარის ამპროლიუმის სიმაღლეების საშუალო სიმაღლიდან (ფართობიდან) საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.3.2). ამპროლიუმის ნიმუშის შემცველობა w მგ/კგ-ში მოცემულია შემდეგი ფორმულით:
სადაც: V = ექსტრაქციული გამხსნელის მოცულობა (ამ თავის პუნქტი 3.8) მლ-ში ამ თავის 5.2-ე პუნქტის შესაბამისად (ანუ 200 მლ); c = ნიმუშის ექსტრაქტის ამპროლიუმის კონცენტრაცია (ამ თავის პუნქტი 5.2) მკგ/მლ-ში; f = განზავების ფაქტორი ამ თავის 5.2-ე პუნქტის შესაბამისად; m = გამოსაკვლევი პორციის წონა გრამებში.
7. შედეგების ვალიდაცია 7.1. იდენტიფიკაცია ანალიტის იდენტურობა შესაძლება დადასტურდეს თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც ხდება ნიმუშის ექსტრაქტის სპექტრებისა (ამ თავის პუნქტი 5.2) და 2.0 მკგ/მლ-ის შემცველობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.7.3) შედარება.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი (5.2) გამდიდრებულია შესაბამისი რაოდენობის საკალიბრო ხსნარის დამატებით (3.7.3). ამპროლიუმის დამატებული რაოდენობა უნდა იყოს ამპროლიუმის რაოდენობის მსგავსი, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ ამპროლიუმის პიკის სიმაღლე. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს ამპროლიუმის მწვერვალის თავდაპირველი სიგანის არაგამდიდრებული ნიმუშის ექსტრაქტის ± 10%-ში ფარგლებში.
7.1.2. გამოვლენა დიოდური – მატრიცის გამოყენებით შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით: ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბსორბაციის (შეწოვის) ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, იდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცის გამოვლენისთვის ეს ჩვეულებრივ ±2 ნმ-ის ფარგლებშია; ბ) 210-დან 320 ნმ-მდე, ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100% ფარდობითი აბსორბაციის (შეწოვის) დიაპაზონში. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული ანალიტის შთანთქმის 15%-ს; გ) 210-დან 320 ნმ-მდე, ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15% -ს. თუ ამ კრიტერიუმებიდან რომელიმე არ სრულდება, ანალიტის არსებობა არ დასტურდება.
7.2. განმეორებადობა ერთი და იმავე ნიმუშზე ჩატარებული ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს: – 15%-ს უფრო მაღალ მნიშვნელობასთან შედარებისას ამპროლიუმის 25 მგ/კგ-დან 500 მგ/კგ-მდე შემცველობისთვის; – 75 მგ/კგ-ს ამპროლიუმის 500 მგ/კგ-დან 1 000 მგ/კგ-მდე შემცველობისთვის; – 7,5%-ს უფრო მაღალ შემცველობასთან შედარებისას ამპროლიუმის 1000 მგ/კგ შემცველობისთვის.
7.3. აღდგენა გამდიდრებული საკონტროლო ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 90%.
8. ერთობლივი კვლევის შედეგები მოეწყო EC-ს ერთობლივი კვლევა, რომელშიც გამოკვლეულ იქნა ფრინველის სამი საკვები (ნიმუში 1-3), ცხოველის ერთი მინერალური საკვები (ნიმუში 4) და ერთი პრემიქსი (ნიმუში 5). შედეგები მოცემულია ქვემოთ ცხრილში:
L = ლაბორატორიების რაოდენობა n = ერთეულების მნიშვნელობების რაოდენობა Sr = განმეორებადობის სტანდარტული გადახრა CVr = განმეორებადობის ვარიაციის კოეფიციენტი SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა CV R = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი.
9. დაკვირვება 9.1. თუ ნიმუში შეიცავს თიამინს, თიამინის პიკი ქრომატოგრამაში ჩნდება ამპროლიუმის პიკამდე ცოტა ხნით ადრე. ამ მეთოდის გამოყენებით უნდა განცალკავდეს ამპროლიუმი და თიამინი. თუ ამპროლიუმი და თიამინი არ განცალკავდება ამ მეთოდში გამოყენებული სვეტით (ამ თავის პუნქტი 4.1.1), მობილური ფაზის 50%-მდე აცეტონიტრილის პორცია ჩაანაცვლეთ მეთანოლით.
9.2. ბრიტანული ფარმაკოპეის თანახმად, ამპროლიუმის ხსნარის სპექტრი (c = 0,02 მოლი/ლიტრი) მარილმჟავაში (c = 0,1 მოლი/ლიტრი) აჩვენებს მაქსიმუმს 246 ნმ და 262 ნმ. შთანთქმის რაოდენობა უნდა იყოს 0,84 246 ნმ და 0,80 262 ნმ-ზე.
9.3. ექსტრაქტი ყოველთვის უნდა განზავდეს მობილური ფაზით, რადგან წინააღმდეგ შემთხვევაში ამპროლიუმის პიკის შეკავების დრომ შეიძლება მნიშვნელოვნად გადაინაცვლოს, იონურ სიმტკიცეში ცვლილების გამო.
თავი IV კარბადოქსის განსაზღვრა
მეთილის 3 – (2-ქინოქსალინილმეთილენ) კარბაზატი N1, N4 -დიოქსიდი
1. მიზანი და გამოყენების ფარგლები ეს მეთოდი საშუალებას იძლევა განვსაზღვროთ კარბადოქსის შემცველობა ცხოველის საკვებში, პრემიქსებსა და პრეპარატებში. გამოვლენის ზღვარი შეადგენს 1 მგ/კგ-ს. რაოდენობრივი შეფასების ზღვარი კი – 5 მგ/კგ-ს.
2. გამოყენების პრინციპი ნიმუშის გათანაბრება ხდება წყლით და ხდება მისი ექსტრაჰირება მეთანოლ-აცეტონიტრილით. ცხოველის საკვებივის, გაფილტრული ექსტრაქტის ალიქვოტის პორცია ექვემდებარება გაწმენდას ალუმინის ოქსიდის სვეტზე. პრემიქსებისა და პრეპარატებისათვის გაფილტრული ალიქვოტის პორცია განზავებულია შესაბამის კონცენტრაციამდე წყალთან, მეთანოლთან და აცეტ-ონიტრილთან. კარბადოქსის შემცველობა განისაზღვრება შებრუნებული ფაზის მაღალი ეფექტურობის თხევადი ქრომატოგრაფიით (HPLC), UV დეტექტორით.
3. რეაგენტები 3.1. მეთანოლი. 3.2. აცეტონიტრილი, HPLC ხარისხის ეკვივალენტური. 3.3. ძმარმჟავა, w = 100%. 3.4. ალუმინის ოქსიდი: ნეიტრალური, აქტივობის I ხარისხი. 3.5. მეთანოლ-აცეტონიტრილი 1 + 1 (v + v). შეურიეთ 500 მლ მეთანოლი (ამ თავის პუნქტი 3.1) 500 მლ აცეტონიტრილთან (ამ თავის პუნქტი 3.2). 3.6. ძმარმჟავა, σ = 10%. წყლით განაზავეთ 10 მლ ძმარმჟავა (ამ თავის პუნქტი 3.3) 100 მლ-მდე. 3.7. ნატრიუმის აცეტატი. 3.8. წყალი, HPLC ხარისხის ეკვივალენტური. 3.9. აცეტატის ბუფერული ხსნარი, c = 0,01 მოლი/ლიტრი, pH = 6,0. გახსენით 0,82 გ ნატრიუმის აცეტატი (ამ თავის პუნქტი 3.7) 700 მლ წყალში (ამ თავის პუნქტი 3.8) და pH შეცვალეთ 6,0-მდე ძმარმჟავით (ამ თავის პუნქტი 3.6). გადაიტანეთ 1 000 მლ გრადირებულ კოლბაში, შეავსეთ ნიშნულამდე წყლით (ამ თავის პუნქტი 3.8) და აურიეთ. 3.10. მობილური ფაზა HPLC-სთვის. აურიეთ 825 მლ აცეტატის ბუფერული ხსნარი (ამ თავის პუნქტი 3.9) 175 მლ აცეტონიტრილთან (ამ თავის პუნქტი 3.2). გაფილტრეთ 0,22 მკმ ფილტრის საშუალებით (ამ თავის პუნქტი 4.5) და გაანაწილეთ ხსნარი (მაგ., ულტრასონიფიკაციით 10 წუთის განმავლობაში). 3.11. სტანდარტული ნივთიერება. სუფთა კარბადოქსი: მეთილის 3 – (2-ქინოქსალინილმეთილენის) კარბაზატი N1, N4 – დიოქსიდი, E 850. 3.11.1. კარბადოქსის საწყისი სტანდარტული ხსნარი, 100 მკგ/მლ (იხ. ამ თავის შენიშვნა 5. პროცედურა): აწონეთ 0,1 მგ სიზუსტით, 25 მგ კარბადოქსის სტანდარტული ნივთიერება (ამ თავის პუნქტი 3.11) 250მლ გრადირებულ კოლბაში. გახსენით მეთანოლ-აცეტონიტრილში (ამ თავის პუნქტი 3.5) ულტრასონიფიკაციით (ამ თავის პუნქტი 4.7). ულტრაბგერითი დამუშავების შემდეგ ხსნარი მიიყვანეთ ოთახის ტემპერატურამდე, შეავსეთ ნიშნულამდე მეთანოლ-აცეტონიტრილით (ამ თავის პუნქტი 3.5) და აურიეთ. შეფუთეთ კოლბა ალუმინის ფოლგით ან გამოიყენეთ ქარვის მინის ჭურჭელი და შეინახეთ მაცივარში. ≤ 4oC ტემპერატურაზე ხსნარი სტაბილურია 1 თვის განმავლობაში. 3.11.2. საკალიბრო ხსნარები 2,0, 5,0, 10,0 და 20,0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.11.1) გადაიტანეთ 100 მლ გრადირებული კოლბების სერიაში. დაამატეთ 30 მლ წყალი, შეავსეთ ნიშნულამდე მეთანოლ-აცეტონიტრილით (ამ თავის პუნქტი 3.5) და აურიეთ. შეფუთეთ კოლბები ალუმინის ფოლგით. ეს ხსნარები შეესაბამება 2,0, 5,0, 10,0 და 20,0 მკგ/მლ კარბადოქსს შესაბამისად. აღნიშნული ხსნარები, გამოყენებამდე უნდა იყოს ახალი მომზადებული.
შენიშვნა: ცხოველის საკვებში კარბადოქსის დასადგენად, რომელიც შეიცავს 10 მგ/კგ-ზე ნაკლებს, უნდა მომზადდეს საკალიბრო ხსნარები 2,0 მკგ/მლ-ზე ნაკლები კონცენტრაციით. 3.12. წყლის – [მეთანოლ-აცეტონიტრილის] (ამ თავის პუნქტი 3.5) ნარევი, 300 + 700 (v + v). შეურიეთ 300 მლ წყალი 700 მლ მეთანოლ-აცეტონიტრილის ნარევს (ამ თავის პუნქტი 3.5).
4. აპარატურა 4.1. ლაბორატორიული სადღვები ან მაგნიტური სათქვეფელა. 4.2. მინის ბოჭკოვანი ფილტრის ქაღალდი (Whatman GF/A ან ეკვივალენტი). 4.3. მინის სვეტი (სიგრძე 300-დან 400 მმ, შიდა დიამეტრი დაახლოებით 10მმ) აგლომერირებული მინის ფრიტით და გამყვანი სარქველით. შენიშვნა: ასევე შეიძლება გამოყენებულ იქნეს მინის სვეტი, რომელზეც დამონტაჟებულია საცობი ან მინის სვეტი, რომელსაც აქვს დახურული ბოლო; ამ შემთხვევაში, ქვედა ბოლოში თავსდება მინაბოჭკოვანი ბამბის საცობი, რომელიც იტკეპნება (იპრესება) მინის ჯოხით. 4.4. HPLC მოწყობილობა ინჯექტირების სისტემით, რომელიც შესაფერისია 20 მკლ ინჯექტირების მოცულობებისთვის. 4.4.1. თხევადი ქრომატოგრაფიული სვეტი: 300 მმ × 4 მმ, C18, 10 მკმ შეფუთვა ან ეკვივალენტი 4.4.2. UV დეტექტორი ტალღის ცვალებადი რეგულირებით ან დიოდური მატრიცის დეტექტორით, რომელიც მუშაობს 225-დან 400 ნმ-მდე დიაპაზონში. 4.5. მემბრანული ფილტრები, 0.22 მკმ 4.6. მემბრანული ფილტრები, 0,45 მკმ 4.7. ულტრაბგერითი აბაზანა.
5. გამოყენების პროცედურა შენიშვნა: კარბადოქსი მგრძნობიარეა სინათლის მიმართ. ჩაატარეთ ყველა პროცედურა სუსტი შუქის ქვეშ ან გამოიყენეთ ქარვის მინის ჭურჭელი ან ალუმინის ფოლგაში გახვეული მინის ჭურჭელი.
5.1. ზოგადი პრინციპი 5.1.1. ცხოველის საკონტროლო საკვები აღდგენითი ტესტის (ამ თავის პუნქტი 5.1.2) შესასრულებლად უნდა მოხდეს ცხოველის საკონტროლო საკვების გამოკვლევა, რათა შემოწმდეს კარბადოქსი და ხელის შემშლელი ნივთიერებების არარსებობა. ცხოველის საკონტროლო საკვები უნდა იყოს ნიმუშის მსგავსი და კარბადოქსი ან ხელის შემშლელი ნივთიერებები არ უნდა გამოვლინდეს.
5.1.2. აღდგენითი ტესტი აღდგენითი ტესტი უნდა ჩატარდეს ცხოველის საკონტროლო საკვების (ამ თავის პუნქტი 5.1.1) გამოკვლევით, რომელიც გამდიდრებულია კარბადოქსის იმ რაოდენობის დამატებით, რომელიც ნიმუშში არსებულის ანალოგიურია. 50 მგ/კგ დონემდე გამდიდრებისთვის, გადაიტანეთ 5.0 მლ საწყისი სტანდარტული ხსნარი (ამ თავის პუნქტი 3.11.1) 200 მლ კონუსური ფორმის კოლბაში. აორთქლეთ ხსნარი დაახლოებით 0,5 მლ აზოტის ნაკადში. დაამატეთ 10 გრ ცხოველის საკონტროლო საკვები, აურიეთ და გააჩერეთ 10 წთ ვიდრე გადახვოდოდეთ ექსტრაქციის ეტაპზე (ამ თავის პუნქტი 5.2). ალტერნატიულად, თუ ნიმუშის მსგავსი ტიპის ცხოველის საკონტროლო საკვები არ არის ხელმისაწვდომი (იხ. ამ თავის პუნქტი 5.1.1), აღდგენითი ტესტი შეიძლება ჩატარდეს სტანდარტული დამატების მეთოდით. ამ შემთხვევაში, ხდება გამოსაკვლევი ნიმუშის გამდიდრება კარბადოქსის რაოდენობით, ანალოგიურად, როგორც ეს უკვე წარმოდგენილია ნიმუშში. ხდება ამ ნიმუშის გამოკვლევა გაუმდიდრებელ ნიმუშთან ერთად და აღდგენის გამოანგარიშება შესაძლებელია გამოკლებით.
5.2. ექსტრაჰირება
5.2.1. ცხოველის საკვები აწონეთ 0,01 გ სიზუსტით, 10 გ ნიმუში და გადაიტანეთ 200 მლ კონუსური ფორმის კოლბაში. დაამატეთ 15,0 მლ წყალი, აურიეთ და გაათანაბრეთ 5 წთ. დაამატეთ 35,0 მლ მეთანოლ-აცეტონიტრილი (ამ თავის პუნქტი 3.5), გაუკეთეთ საცობი და შეანჯღრიეთ 30 წთ სადღვებზე ან ურიეთ მაგნიტურ სადღვებელაზე (ამ თავის პუნქტი 4.1). ხსნარი გაფილტრეთ მინის ბოჭკოვანი ფილტრის ქაღალდის საშუალებით (ამ თავის პუნქტი 4.2). შეინახეთ ეს ხსნარი გაწმენდის ეტაპისთვის (ამ თავის პუნქტი 5.3).
5.2.2. პრემიქსები (0, 1% – 2, 0%) აწონეთ 0,001 გ სიზუსტით 1 გ დაუფქვავი ნიმუში და გადაიტანეთ 200 მლ კონუსური ფორმის კოლბაში. დაამატეთ 15,0 მლ წყალი, აურიეთ და გაათანაბრეთ 5 წთ დაამატეთ 35,0 მლ მეთანოლ-აცეტონიტრილი (ამ თავის პუნქტი 3.5), გაუკეთეთ საცობი და შეანჯღრიეთ 30 წთ სადღვებზე ან ურიეთ მაგნიტურ სადღვებელაზე (ამ თავის პუნქტი 4.1). ხსნარი გაფილტრეთ მინის ბოჭკოვანი ფილტრის ქაღალდის საშუალებით (ამ თავის პუნქტი 4.2). პიპეტით გადაიტანეთ ფილტრატის ალიქვოტი 50-მლ-იან დაკალიბრებულ კოლბაში. დაამატეთ 15,0 მლ წყალი, შეავსეთ მეთანოლ-აცეტონიტრილით (ამ თავის პუნქტი 3.5) და აურიეთ. კარბადოქსის საბოლოო ხსნარის კონცენტრაცია უნდა იყოს დაახლოებით 10 მკგ/მლ. მოახდინეთ ალიქვოტის გაფილტვრა 0,45 მკმ ფილტრის საშუალებით (ამ თავის პუნქტი 4.6).
გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.4).
5.2.3. პრეპარატები (> 2%) აწონეთ 0,001 გ სიზუსტით, 0,2 გ დაუფქვავი ნიმუში და გადაიტანეთ 250 მლ კონუსური ფორმის კოლბაში. დაამატეთ 45,0 მლ წყალი, აურიეთ და გაათანაბრეთ 5 წთ დაამატეთ 105,0 მლ მეთანოლ-აცეტონიტრილი (ამ თავის პუნქტი 3.5), გაუკეთეთ საცობი და მოახდინეთ ჰომოგენიზაცია.
მოახდინეთ ნიმუშის დამუშავება ულტრაბგერებით (ამ თავის პუნქტი 4.7) 15 წთ-ით, რის შემდეგ 15 წთ შეანჯღრიეთ ან მოურიეთ (ამ თავის პუნქტი 4.1). ხსნარი გაფილტრეთ მინის ბოჭკოვანი ფილტრის ქაღალდით (ამ თავის პუნქტი 4.2).
განაზავეთ ფილტრატის ალიქვოტი წყლის-მეთანოლ-აცეტი-ონიტრილის ნარევით (ამ თავის პუნქტი 3.12) 10-15 მკგ/მლ კარბადოქსის საბოლოო კონცენტრაციამდე (10-იანი მომზადებისთვის, განზავების კოეფიციენტი არის 10). ალიქვოტის ფილტრაცია ხდება 0,45 მკმ ფილტრით (ამ თავის პუნქტი 4.6).
გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.4).
5.3. განწმენდა (გასუფთავება) 5.3.1. ალუმინის ოქსიდის სვეტის მომზადება აწონეთ 4 გ ალუმინის ოქსიდი (ამ თავის პუნქტი 3.4) და გადაიტანეთ მინის სვეტზე (ამ თავის პუნქტი 4.3).
5.3.2. ნიმუშის პურიფიკაცია დაიტანეთ 15 მლ გაფილტრული ექსტრაქტი (ამ თავის პუნქტი 5.2.1) ალუმინის ოქსიდის სვეტზე და მოაცილეთ პირველი 2 მლ ელუატი. შეაგროვეთ მომდევნო 5 მლ და გაფილტრეთ ალიქვოტი 0,45 მკმ ფილტრით (ამ თავის პუნქტი 4.6). გადადით HPLC განსაზღვრაზე (ამ თავის პუნქტი 5.4).
5.4. HPLC განსაზღვრა 5.4.1. პარამეტრები სახელმძღვანელოდ შემოთავაზებულია შემდეგი პირობები; შესაძლებელია სხვა პირობების გამოყენება იმ პირობით, თუ ისინი ეკვივალენტურ შედეგებს იძლევიან:
შეამოწმეთ ქრომატოგრაფიული სისტემის სტაბილურობა, რამდენჯერმე მოახდინეთ საკალიბრო ხსნარი შეყვანა (ინჯექტირება) (ამ თავის პუნქტი 3.11.2), რომელიც შეიცავს 5,0 მკგ/მლ, სანამ არ მიიღწევა პიკის მუდმივი სიმაღლეები (ფართობები) და შენარჩუნების დრო.
5.4.2. საკალიბრო მრუდი რამდენჯერმე მოახდინეთ თითოეული საკალიბრო ხსნარის ინჯექტირება (ამ თავის პუნქტი 3.11.2) და გაზომეთ პიკის სიმაღლეები (ფართობები) თითოეული კონცენტრაციისთვის. შეადგინეთ საკალიბრო მრუდი, საკალიბრო ხსნარების საშუალო პიკის სიმაღლეების (ფართობების), როგორც ორდინატას და შესაბამისი კონცენტრაციების მკგ/მლ-ში, როგორც აბცისასი.
5.4.3. ნიმუშის ხსნარი რამდენჯერმე მოახდინეთ ნიმუშის ექსტრაქტის ინჯექტირება [(ამ თავის პუნქტი 5.3.2) ცხოველის საკვებისთვის, (ამ თავის პუნქტი 5.2.2) პრემიქსებისა და (ამ თავის პუნქტი 5.2.3) პრეპარატებისთვის] და განსაზღვრეთ კარბადოქსის მწვერვალების საშუალო პიკის სიმაღლე (ფართობი).
6. შედეგების ფორმულირება ნიმუშის ხსნარის კარბადოქსის პიკების საშუალო სიმაღლიდან (ფართობიდან) განსაზღვრეთ კარბადოქსის ნიმუშის კონცენტრაცია მკგ/მლ-ში საკალიბრო მრუდის მითითებით (ამ თავის პუნქტი 5.4.2).
6.1. ცხოველის საკვები კარბადოქსის შემცველობა (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ექსტრაქტის კარბადოქსის კონცენტრაცია (ამ თავის პუნქტი 5.3.2) მკგ/მლ V1 = ექსტრაჰირების მოცულობა მლ (მაგ., 50) m = გამოსაკვლევი პორციის წონა გ-ში.
6.2. პრემიქსები და პრეპარატები
კარბადოქსის შემცველობა (მგ/კგ) მოცემულია შემდეგი ფორმულით:
სადაც: c = ნიმუშის ექსტრაქტის კარბადოქსის კონცენტრაცია (ამ თავის პუნქტი 5.2.2 ან 5.2.3) მკგ/მლ-ში; V2 = ექსტრაქციის მოცულობა მლ-ში (მაგ., 50 პრემიქსებისთვის; 150 პრეპარატებისთვის); f = განზავების კოეფიციენტი 5.2.2 (პრემიქსები) ან 5.2.3 (პრეპარატები); m = გამოსაკვლევი პორციის წონა გ-ში.
7. შედეგების ვალიდაცია 7.1. იდენტიფიკაცია
ანალიტის იდენტურობა შესაძლება დადასტურდეს თანაქრომატოგრაფიით, ან დიოდური მატრიცის დეტექტორის გამოყენებით, რომლითაც ხდება ნიმუშის ექსტრაქტის სპექტრებისა და 10.0 მკგ/მლ-ის შემცველობის საკალიბრო ხსნარის (ამ თავის პუნქტი 3.11.2) შედარება.
7.1.1. თანაქრომატოგრაფია ნიმუშის ექსტრაქტი გამდიდრებულია შესაბამისი რაოდენობის საკალიბრო ხსნარის დამატებით (ამ თავის პუნქტი 3.11.2). კარბადოქსის დამატებული რაოდენობა უნდა იყოს კარბადოქსის რაოდენობის მსგავსი, რომელიც აღმოჩენილი იქნა ნიმუშის ექსტრაქტში.
დამატებული რაოდენობისა და ექსტრაქტის განზავების გათვალისწინების შემდეგ უნდა გაიზარდოს მხოლოდ კარბადოქსის პიკის სიმაღლე. პიკის სიგანე, მისი მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს თავდაპირველი სიგანის დაახლოებით ±10% ფარგლებში.
7.1.2. გამოვლენა დიოდი – მატრიცის გამოყენებით შედეგების შეფასება ხდება შემდეგი კრიტერიუმებით: ა) ნიმუშისა და სტანდარტული სპექტრის მაქსიმალური აბsორბაციის (შეწოვის) ტალღის სიგრძე, რომელიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, იდენტური უნდა იყოს იმ ფარგლებში, რომელიც განსაზღვრულია გამოვლენის სისტემის გადამწყვეტი სიმძლავრით. დიოდური მატრიცის გამოვლენისთვის ეს ჩვეულებრივ +2 ნმ-ის ფარგლებშია;
ბ) 225-დან 400 ნმ-მდე, ნიმუშისა და სტანდარტის სპექტრები, რომლებიც დაფიქსირებულია პიკის სიმაღლეზე ქრომატოგრამაში, არ უნდა განსხვავდებოდეს სპექტრის ამ ნაწილებისათვის 10%-დან 100%-მდე ფარდობითი აბსორბაციის (შეწოვის) დიაპაზონში. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და როდესაც ყველა დაფიქსირებულ წერტილში ორ სპექტრს შორის გადახრა არ აღემატება სტანდარტული ანალიტის შთანთქმის 15%-ს; გ) 225-დან 400 ნმ-მდე, ნიმუშის ექსტრაქტის მიერ წარმოქმნილი ამსვლელი, მწვერვალისა და ჩამომსვლელი სპექტრი არ უნდა განსხვავდებოდეს ერთმანეთისგან სპექტრის იმ ნაწილებისთვის, რომელიც შედარებითი შთანთქმის 10%-დან 100%-ს შორისაა. ეს კრიტერიუმი სრულდება მაშინ, როდესაც სახეზეა იმავე მაქსიმუმი და, როდესაც ყველა დაფიქსირებულ წერტილში სპექტრებს შორის გადახრა არ აღემატება მწვერვალის სპექტრის აბსორბციის 15%-ს.
თუ ამ კრიტერიუმებიდან რომელიმე არ სრულდება, მაშინ ანალიტის არსებობა არ დასტურდება.
7.2. განმეორებადობა 10 მგ/კგ და მეტი შემცველობისთვის, ერთსა და იმავე ნიმუშზე ორი პარალელური განსაზღვრის შედეგებს შორის სხვაობა არ უნდა აღემატებოდეს 15%-ს უფრო მაღალ შედეგთან შედარებისას.
7.3. აღდგენა გამდიდრებული (საკონტროლო) ნიმუშისთვის აღდგენა სულ მცირე უნდა იყოს 90%.
8. ერთობლივი კვლევის შედეგები ჩატარდა ერთობლივი გამოკვლევა, სადაც 8 ლაბორატორიამ გამოიკვლია 6 ცხოველის საკვები, 4 პრემიქსი და 3 პრეპარატი. თითოეულ ნიმუშზე ჩატარდა განმეორებითი (დუბლიკატი) გამოკვლევა. (ამ ერთობლივი კვლევის შესახებ უფრო დეტალური ინფორმაცია შეგიძლიათ იხილოთ ჟურნალში AOAC, ტომი 71, 1988, გვ. 484-490). შედეგები (გარდა ცდომილებებისა) ნაჩვენებია ქვემოთ:
ცხრილი №1 ცხოველის საკვების ერთობლივი კვლევის შედეგები
ცხრილი №2
პრემიქსებისა და პრეპარატების ერთობლივი კვლევის შედეგები
L = ლაბორატორიების რაოდენობა; n = ერთეულების მნიშვნელობების რაოდენობა; Sr = განმეორებადობის სტანდარტული გადახრა; CVr = განმეორებადობის ვარიაციის კოეფიციენტი; SR = რეპროდუქციულობის (შედეგიანობის) სტანდარტული გადახრა; CVR = რეპროდუქციულობის (შედეგიანობის) ვარიაციის კოეფიციენტი.
1 უნდა მოხდეს ნებისმიერი ნატეხის დაყოფა (საჭიროების შემთხვევაში მათი გაყოფითა და ნიმუშში დაბრუნებით). 2 დაბალი კუთრი წონის მქონე უხეში საკვებისა და ფურაჟის გარდა. 3 დაბალი კუთრი წონის მქონე უხეში საკვებისა და ფურაჟის გარდა. [4] მარცვლეულის, ფქვილის, ბურღულისა და უხეში ფქვილის გასაშრობად, ღუმელს უნდა ჰქონდეს ისეთი თერმული სიმძლავრე, რომ წინასწარ 131°C ტემპერატურაზე დაყენების შემდეგ, უბრუნდებოდეს შესაბამის ტემპერატურას 45 წუთზე ნაკლებ დროში, ერთდროულად გაშრობის მიზნით, ნიმუშების მაქსიმალური რაოდენობის შიგნით მოთავსების შემდეგ. ვენტილაციამ უნდა უზრუნველყოს, რომ მაქსიმალური რაოდენობის შემცველი ხორბლის ნიმუშების, რომლებიც ორი საათის განმავლობაში უნდა გაშრეს, შედეგები განსხვავდებოდეს ოთხი საათის გაშრობის შემდეგ მიღებული შედეგებისგან მინუს 0,15%-ით. [5] თუ ზეთს ან ცხიმს უნდა ჩაუტარდეს შემდგომი ტესტირება ხარისხზე, პემზის ქვის ფრაგმენტები შეცვალეთ მინის ბურთულებით. [6] ჩატარდა Verband Deutscher Landwirtschaftlicher Untersuchungs – und Forschungsanstalten (VDLUFA) Feed-ის სამუშაო ჯგუფის მიერ. [7] ჩატარდა Verband Deutscher Landwirtschaftlicher Untersuchungs – und Forschungsanstalten (VDLUFA) Feed-ის სამუშაო ჯგუფის მიერ. [8] შეიძლება გამოყენებულ იქნეს გადახარშვის სხვა მეთოდები, თუ აჩვენეს, რომ მათ აქვთ მსგავსი შედეგები (მაგალითად, მიკროტალღური წნევით გადახარშვა). [9] მწვანე საკვები (ახალი ან გამომშრალი) შეიძლება შეიცავდეს დიდი რაოდენობით მცენარეულ კრემნიუმის დიოქსიდს (silica), რომელსაც შეუძლია შეინარჩუნოს კვალი ელემენტები და უნდა იქნეს მოცილებული. ცხოველის საკვების ამ ნიმუშებისთვის, შესაბამისად, უნდა შესრულდეს შემდეგი მოდიფიცირებული პროცედურა. განხორციელეთ ფილტრაციიის პროცედურა ამ თავის 5.1.1.1 პუნქტის შესაბამისად, უხსნადი ნაშთის (ნარჩენების) შემცველი ფილტრის ქაღალდი ორჯერ გარეცხეთ მდუღარე წყალში და მოათავსეთ იგი კვარცის ან პლატინის თიგელში. მოახდინეთ აალება მუფელურ ღუმელში (ამ თავის პუნქტი 4.1) 550oC-ზე დაბალ ტემპერატურაზე, სანამ კარბონატული მასალა მთლიანად არ გაქრება. გააგრილეთ, დაამატეთ რამდენიმე წვეთი წყალი, რასაც უნდა მოჰყვეს 10-დან 15 მლ-მდე ფტორწყალბადმჟავას (ამ თავის პუნქტი 3.4) დამატება და ააორთქლეთ გაშრობამდე დაახლოებით 150oC-ზე. თუ კრემნიუმის დიოქსიდს (silica) დარჩება ნაშთში (ნარჩენებში), გადაანაწილეთ იგი რამდენიმე მილილიტრ ფტორწყალბადმჟავაში (ამ თავის პუნქტი 3.4) და აორთქლეთ გაშრობამდე. დაამატეთ ხუთი წვეთი გოგირდმჟავა (ამ თავის პუნქტი 3.5) და გააცხელეთ მანამ, სანამ არ გამოიყოფა თეთრი ორთქლი. 5 მლ 6 მოლი/ლ მარილმჟავას (ამ თავის პუნქტი 3.2) და დაახლოებით 30 მლ წყლის დამატების შემდეგ, გაათბეთ, გაფილტრეთ ხსნარი 250 მლ მოცულობის კოლბაში და შეავსეთ წყლით (HCl კონცენტრაცია დაახლოებით 0,5 მოლ/ლ)). შემდეგ გადადით განსაზღვრაზე ამ თავის 5.1.2 პუნქტის მიხედვით. [10] TEF (=ტოქსიკურობის ეკვივალენტურობის ფაქტორი) ცხრილი PCDD-ებისთვის, PCDF!ებისთვის და დიოქსინის მსგავსი PCB-ებისთვის: ადამიანის რისკის შეფასების WHO-TEF-ები ეფუძნება ჟენევაში, 2005 წლის ივნისში ჩატარებულ ჯანმრთელობის მსოფლიო ორგანიზაციის (WHO) – ქიმიური უსაფრთხოების საერთაშორისო პროგრამის (IPCS) ექსპერტის შეხვედრის დასკვნებს. (მარტინ ვან დენ ბერგ და სხვ., 2005 წლის მსოფლიო ჯანდაცვის ორგანიზაციის მიერ ადამიანისა და ძუძუმწოვრების ტოქსიკურობის ეკვივალენტურობის ფაქტორის გადაფასება დიოქსინებისა და დიოქსინის მსგავსი ნაერთებისთვის. ტოქსიკოლოგიური მეცნიერებები 93 (2), 223-241 (2006)) (იხილეთ ცხრილი შენიშვნებში). 11 „განუსაზღვრელობის განსაზღვრის შესახებ სახელმძღვანელო დოკუმენტი ლაბორატორიებისათვის, რომლებიც ახორციელებენ PCDD/F და PCB გამოკვლევას იზოტოპის განზავებითი მას-სპექტომეტრიის გამოყენებით, (http://ec.europa.eu/food/safety/animal-feed_en). 12 განმეორებითი (დუბლიკატი) გამოკვლევა: ინტერესის ანალიტის ცალკეული გამოკვლევა იმავე ჰომოგენიზებული ნიმუშის მეორე ალიქვოტის გამოყენებით. ზოგადად, გამოიყენება დუბლიკატი გამოკვლევისათვის ამ წესის დანართის №2 თავი III -ის მე-3 პუნქტით განსაზღვრული მოთხოვნები. თუმცა შესაბამისი ანალიტისათვის 13C ნიშანდებული შიდა სტანდარტის გამოყენების მეთოდებისთვის, განმეორებითი (დუბლიკატი) გამოკვლევა საჭიროა მხოლოდ იმ შემთხვევაში, თუ პირველი განსაზღვრის შედეგი არ არის შესაბამისობაში. განმეორებითი (დუბლიკატი) გამოკვლევა აუცილებელია იმისთვის, რომ გამოირიცხოს ნიმუშების შიდა ჯვარედინი დაბინძურება ან შემთხვევითი შერევა. იმ შემთხვევაში, თუ გამოკვლევა განხორციელდება დაბინძურების ინციდენტის დროს, განმეორებითი (დუბლიკატი) გამოკვლევის დადასტურება შეიძლება გამოირიცხოს იმ შემთხვევაში, თუ გამოსაკვლევად შერჩეული ნიმუშები მიკვლევადობით უკავშირდება დაბინძურების ინციდენტს და აღმოჩენილი დონე მნიშვნელოვნად აღემატება მაქსიმალურ შემცველობას (ზღვარს). 13 „ზედა ზღვარის (bound)“ კონცეფცია მოითხოვს რაოდენობრივი ლიმიტის გამოყენებას თითოეული არარაოდენობრივი კონგენერის კონტრიბუციისთვის. „ქვედა ზღვარის“ კონცეფცია მოითხოვს ნულის გამოყენებას თითოეული არარაოდენობრივი კონგენერის კონტრიბუციისთვის. „საშუალო ზღვარის“ კონცეფცია მოითხოვს რაოდენობრივი ლიმიტის ნახევარის გამოყენებას თითოეული არარაოდენობრივი კონგენერის კონტრიბუციის გამოსათვლელად. 14 ზოგადად გამოიყენება განმეორებითი (დუბლიკატი) გამოკვლევისათვის ამ წესის დანართის №2 თავი III-ის მე-2 პუნქტით განსაზღვრული მოთხოვნები. ამასთან, შესაბამისი გამოკვლევისთვის 13C-ნიშანდებული შიდა სტანდარტის გამოყენების მეთოდებისთვის, განმეორებითი (დუბლიკატი) გამოკვლევა საჭიროა მხოლოდ იმ შემთხვევაში, თუ პირველი განსაზღვრის შედეგი არ არის შესაბამისობაში. განმეორებითი (დუბლიკატი) გამოკვლევა აუცილებელია, რომ გამოირიცხოს ნიმუშების შიდა ჯვარედინი დაბინძურება ან შემთხვევითი შერევა. იმ შემთხვევაში, თუ გამოკვლევა განხორციელდება დაბინძურების ინციდენტის დროს, განმეორებითი (დუბლიკატი) გამოკვლევით დადასტურება შეიძლება გამოირიცხოს იმ შემთხვევაში, თუ გამოკვლევისათვის შერჩეული ნიმუშები მიკვლევადობით უკავშირდება დაბინძურების ინციდენტს და აღმოჩენილი ზღვარი მნიშვნელოვნად აღემატება მაქსიმალურ შემცველობას . 15 „ზედა ზღვარის“ კონცეფცია მოითხოვს რაოდენობრივი ლიმიტის გამოყენებას თითოეული რაოდენობრივი კონგენერის TEQ-ში (ტოქსიკური ეკვივალენტობის) კონტრიბუციისთვის. „ქვედა ზღვარის“ კონცეფცია მოითხოვს ნულის გამოყენებას თითოეული არარაოდენობრივი კონგენერის კონტრიბუციისთვის. „საშუალო ზღვარის“ კონცეფცია მოითხოვს TEQ-ში არარაოდენობრივი ლიმიტის ნახევარის გამოყენებას TEQ-ში თითოეული არარაოდენობრივი კონგენერის კონტრიბუციის გამოსათვლელად. 16 იდენტური განმარტება და მოთხოვნები გამოიყენება განმეორებითი (დუბლიკატი) გამოკვლევისას სამოქმედო ზღურბლების კონტროლისთვის, როგორც მე-14 სქოლიოშია, მაქსიმალური შემცველობისათვის (ზღვარისთვის). 17 ბიოანალიტიკური მეთოდები არ არის სპეციფიკური იმ კონგენერებისთვის, რომლებიც შედის TEF – სქემაში. სხვა სტრუქტურულად დაკავშირებული AhR-აქტიური ნაერთები შეიძლება იყოს ნიმუშის ექსტრაქტში, რომლებიც ხელს უწყობენ საერთო რეაგირებას. ამიტომ, ბიოანალიტიკური შედეგები არ შეიძლება იყოს შეფასება, არამედ არის უფრო TEQ შემცველობის მითითება ნიმუშში. 18 „განუსაზღვრელობის განსაზღვრის სახელმძღვანელო დოკუმენტი იმ ლაბორატორიებისთვის, რომლებიც ახორციელებენ PCDD/F და PCB გამოკვლევას იზოტოპების განზავების მასობრივი სპექტრომეტრიის გამოყენებით“ (http://ec.europa.eu/food/safety/animal-feed_en), „LOD და LOQ სახელმძღვანელო დოკუმენტი ცხოველის საკვებსა და სურსათში დამაბინძურებლების გაზომვების შესაფასებლად,“ (http://ec.europa.eu/food/safety/animal-feed_en). 19 არსებული მოთხოვნები ემყარება TEF-ებს, რომლებიც მოცემულია აქ: M. Van den Berg et al, Toxicol Sci 93 (2), 223–241 (2006). 20 ხშირად კონგენერები ელუირდება ერთად, მაგალითად PCB 28/31, PCB 52/69 და PCB 138/163/164. GC/MS-სთვის ასევე უნდა გაითვალისწინოთ შესაძლო ჩარევები (ხელის შეშლა) უფრო ქლორიანი კონგენერების ფრაგმენტებისგან. 21„დამაბინძურებლების სფეროში გაზომვის მიზნით, ცხოველის საკვებსა და სურსათში LOD და LOQ-ის შეფასების სახელმძღვანელო დოკუმენტში“, (http://ec.europa.eu/food/safety/animal-feed_en) აღწერილი პრინციპები უნდა იქნეს დაცული, როდესაც ეს აუცილებელია. 22 რეკომენდებულია, რომ ნიმუშში, დამაბინძურებლის ზღვარზე, გვქონდეს რეაგენტის სუფთა დონის უფრო ნაკლები კონტრიბუცია. ლაბორატორია პასუხისმგებელია, აკონტროლოს სუფთა დონის ვარიაცია, კერძოდ, თუკი ხდება სუფთა დონეების გამოკლება. 23 არსებული მოთხოვნები ემყარება TEF-ებს, რომლებიც გამოქვეყნებულია, დოკუმენტში: M. Van den Berg et al, Toxicol Sci 93 (2), 223–241 (2006). 25 ცხოველის თითოეული სახეობისთვის, რომელზეც გამიზნულია კვლევა, პრაიმერების და სინჯების ჩამონათვალი ხელმისაწვდომია EURL-AP ვებგვერდზე. 26 ფუნქციონალური მასტერმიქსების მაგალითები ხელმისაწვდომია EURL-AP ვებგვერდზე.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
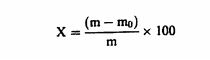
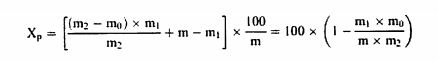
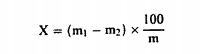
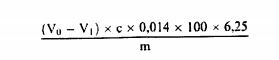
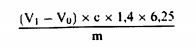
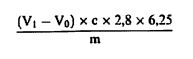
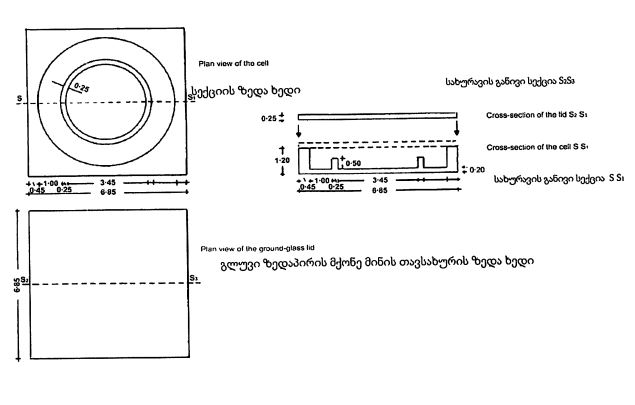
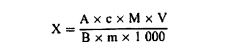
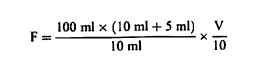
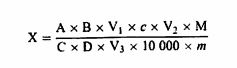

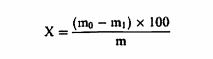

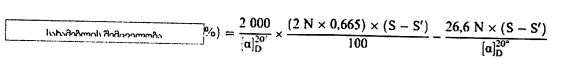
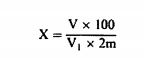
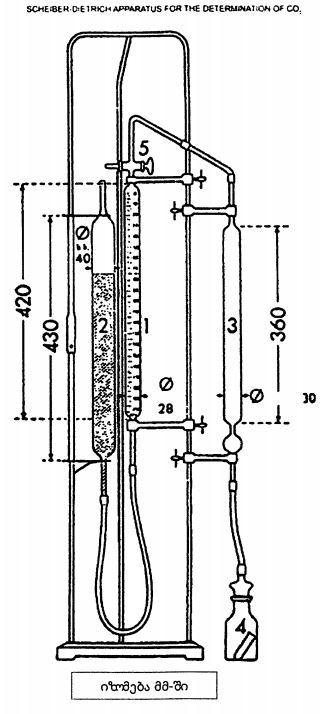
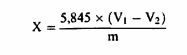


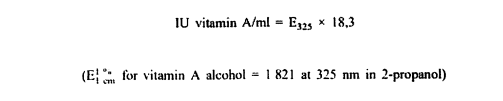
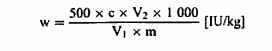
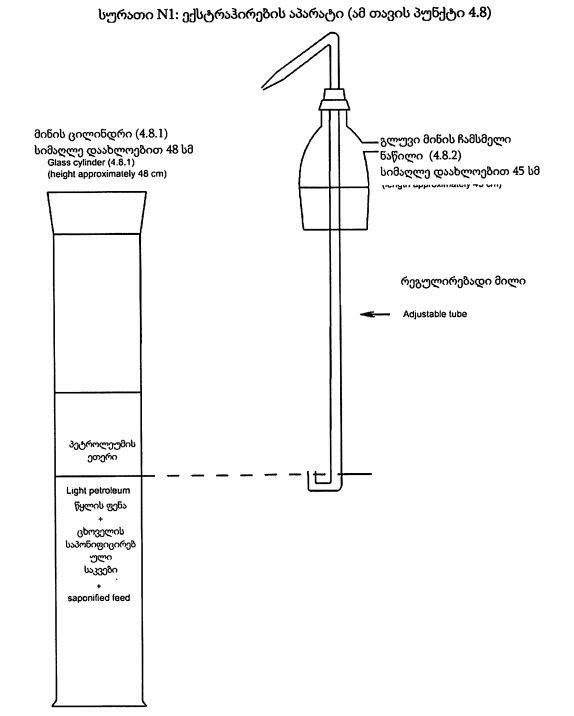

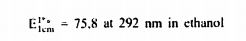
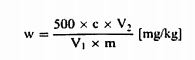
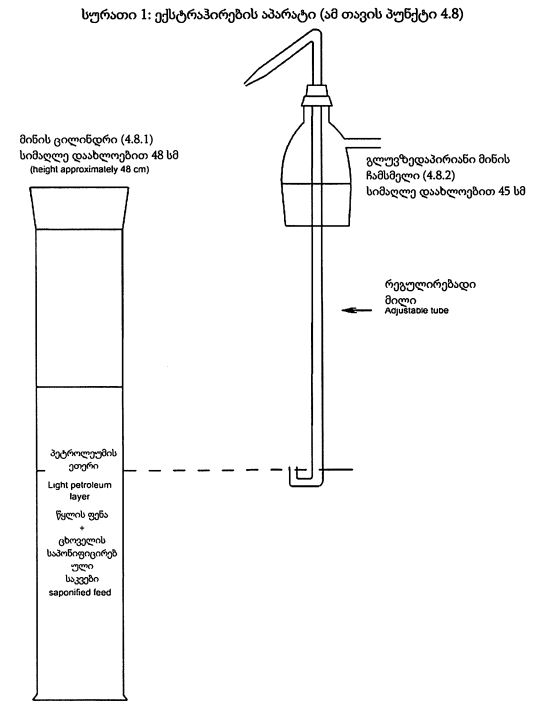
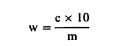

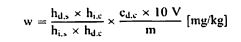
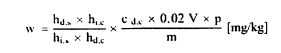
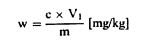
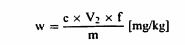

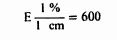
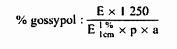
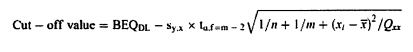
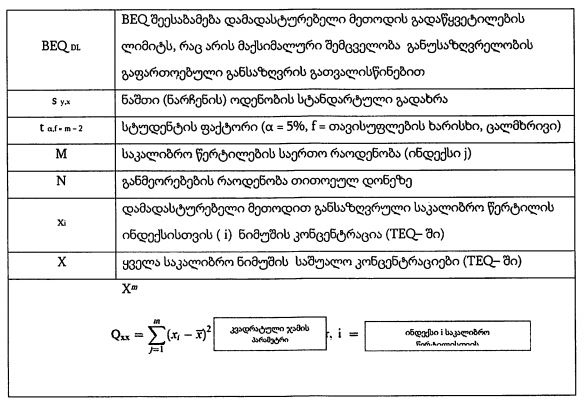
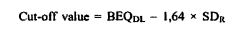
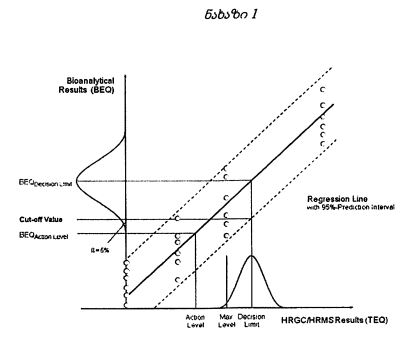
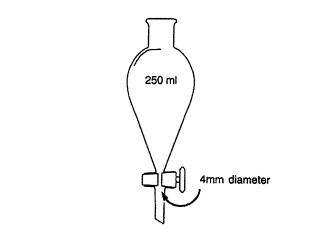

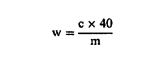

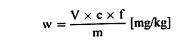
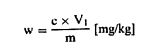
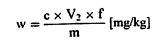
დოკუმენტის კომენტარები