დოკუმენტის სტრუქტურა
განმარტებების დათვალიერება
დაკავშირებული დოკუმენტები
დოკუმენტის მონიშვნები
 უკან დაბრუნება
უკან დაბრუნება | კომისიის 2002 წლის 12 აგვისტოს გადაწყვეტილება, რომელსაც ძალაში შეჰყავს საბჭოს დირექტივა 96/23/EC ანალიზის მეთოდების გამოყენებისა და შედეგების ინტერპრეტაციის შესახებ | |
|---|---|
| დოკუმენტის ნომერი | 96/23/EС |
| დოკუმენტის მიმღები | ევროპის გაერთიანებების კომისია |
| მიღების თარიღი | 12/08/2002 |
| დოკუმენტის ტიპი | გადაწყვეტილება |
| გამოქვეყნების წყარო, თარიღი | ვებგვერდი, 30/03/2017 |
| სარეგისტრაციო კოდი | |
კომისია
კომისიის 2002 წლის 12 აგვისტოს
გადაწყვეტილება,
რომელსაც ძალაში შეჰყავს საბჭოს დირექტივა 96/23/EC ანალიზის მეთოდების გამოყენებისა და შედეგების ინტერპრეტაციის შესახებ
(შეტყობინება გაკეთებულია დოკუმენტის ნომრით C(2002) 3044)
(ტექსტი შეესაბამება ევროპის ეკონომიკურ სივრცეს)
(2002/657/EC)
(აქტები, რომელთა გამოქვეყნებაც სავალდებულო არ არის)
ევროპის გაერთიანებების კომისიამ,
ევროპის გაერთიანების დამფუძნებელი ხელშეკრულების გათვალისწინებით,
საბჭოს 1996 წლის 29 აპრილის დირექტივის 96/23/EC გათვალისწინებით, რომელიც ეხება ცოცხალ ცხოველებსა და ცხოველური წარმოშობის პროდუქტებში გარკვეული ნივთიერებებისა და მათი ნარჩენების კონტროლს და რომელიც აუქმებს დირექტივებს 85/358/EEC და 86/469/EEC და გადაწყვეტილებებს 89/187/EEC და 91/664/EEC1 კერძოდ, აღნიშნული დირექტივის მე-15(1) მუხლის მეორე ქვეაბზაცის გათვალისწინებით,
ვინაიდან:
1) ცხოველური წარმოშობის პროდუქტებში ნარჩენების არსებობა საყურადღებო საკითხია საზოგადოებრივი ჯანმრთელობის თვალსაზრისით.
2) კომისიის 1998 წლის 23 თებერვლის გადაწყვეტილება 98/179/EC რომელიც განსაზღვრავს ცოცხალ ცხოველებსა და ცხოველური წარმოშობის პროდუქტებში გარკვეული ნივთიერებებისა და მათი ნარჩენების კონტროლის მიზნით სინჯების ოფიციალურად აღების დეტალურ წესებს2, ითვალისწინებს სინჯების ანალიზის ჩატარებას მხოლოდ კომპეტენტური სახელმწიფო ორგანოს მიერ ნარჩენების სახელმწიფო კონტროლის მიზნით აღიარებულ ლაბორატორიებში.
3) აუცილებელია ნარჩენების სახელმწიფო კონტროლის მიზნით აღიარებული ლაბორატორიებიდან მიღებული ანალიზის შედეგების ხარისხისა და შედარებადობის უზრუნველყოფა. აღნიშნული მიღწეულ უნდა იქნეს ხარისხის უზრუნველყოფის სისტემების გამოყენებით, კერძოდ, იმ მეთოდების გამოყენებით, რომლებიც დამტკიცებულია საერთო პროცედურებისა და სამუშაო კრიტერიუმების შესაბამისად, ასევე საერთო სტანდარტებთან ან ერთობლივად შეთანხმებულ სტანდარტებთან შესაბამისობის კონტროლის უზრუნველყოფით.
4) საკვები პროდუქტების სახელმწიფო კონტროლის დამატებითი ღონისძიებების შესახებ საბჭოს 1993 წლის 29 ოქტომბრის დირექტივა 93/99/EEC და გადაწყვეტილება 98/179/EC3 მოითხოვს, რომ 2002 წლის იანვრიდან, სახელწიფო კონტროლის განმახორციელებელმა ლაბორატორიებმა აკრედიტაცია გაიარონ ISO 17025(1)-ის მიხედვით. გადაწყვეტილების 98/179/EC თანახმად, აღიარებული ლაბორატორიებს მოეთხოვებათ მონაწილეობა მიიღონ საერთაშორისოდ აღიარებულ გარეშე ხარისხის კონტროლის შეფასებისა და აკრედიტაციის სქემაში. ამასთანავე, აღიარებულმა ლაბორატორიებმა თავიანთი კომპეტენცია უნდა დაამტკიცონ შესაბამისი კვალიფიკაციის ფლობის შესამოწმებელ სქემებში რეგულარული და წარმატებული მონაწილეობით, რომლებიც აღიარებულია ან ორგანიზებულია ეროვნული ან გაერთიანების რეფერალური ლაბორატორიების მიერ.
5) კოორდინაციის გაღრმავების მიზნით, დირექტივის 96/23/EC შესაბამისად, ფუნქციონირებს გაერთიანების რეფერალური ლაბორატორიების, ეროვნული რეფერალური ლაბორატორიებისა და ეროვნული მაკონტროლებელი ლაბორატორიების ქსელი.
6) დირექტივის 96/23/EC მიღების შემდგომ ანალიტიკურ ქიმიაში მიღწეული პროგრესის შედეგად რუტინული მეთოდებისა და ეტალონური მეთოდების ცნება ჩაანაცვლა კრიტერიუმებზე დაფუძნებულმა მიდგომებმა, რომლებიც განსაზღვრავს სამუშაო კრიტერიუმებს, სკრინინგისა და დამადასტურებელი მეთოდების დამტკიცების პროცედურებს.
7) აუცილებელია სახელმწიფო კონტროლის განმახორციელებელი ლაბორატორიების მიერ ჩატარებული შედეგების ინტერპრეტაციისათვის საერთო კრიტერიუმების განსაზღვრა, რათა უზრუნველყოფილი იქნეს დირექტივის 96/23/EC ჰარმონიზებული იმპლემენტაცია.
8) აუცილებელია უზრუნველყოფილი იქნეს ანალიტიკური მეთოდის მინიმალური აუცილებელი სამუშაო ზღვრების (MRPL) ეტაპობრივი დადგენა იმ ნივთიერებებისთვის, რომელთათვისაც დაწესებული არ არის რაიმე დაშვებული ზღვარი, კერძოდ, იმ ნივთიერებებისათვის, რომელთა გამოყენებაც არ არის ნებადართული, ან სპეციალურად არის აკრძალული გაერთიანების ტერიტორიაზე – რათა უზრუნველყოფილი იქნეს დირექტივის 96/23/EC ჰარმონიზებული იმპლემენტაცია.
9) კომისიის 1990 წლის 26 სექტემბრის გადაწყვეტილება 90/515/EEC, რომელიც განსაზღვრავს მძიმე მეტალისა და დარიშხანის აღმოჩენის მეთოდებს4, კომისიის 1993 წლის 14 მაისის გადაწყვეტილება 93/256/EEC, რომელიც განსაზღვრავს იმ ნივთიერებების ნარჩენების აღმოჩენის მეთოდებს, რომლებსაც გააჩნიათ ჰორმონალური ან თიროსტატიკური მოქმედება5 და კომისიის 1993 წლის 15 აპრილის გადაწყვეტილება 93/257/EEC, რომელიც განსაზღვრავს ნარჩენების აღმოჩენასთან დაკავშირებულ ეტალონურ მეთოდებს6 და ეროვნული რეფერალური ლაბორატორიების ნუსხას, რომელშიც ბოლოს ცვლილებები შეტანილი იქნა გადაწყვეტილებით 98/536/EC7, გადახედილ იქნა სამეცნიერო და ტექნიკურ სფეროში არსებული განვითარების გათვალისწინებით, რის შედეგადაც აღნიშნული აქტები აღმოჩნდა მოძველებული მათი მასშტაბებისა და დებულებების თვალსაზრისით და შესაბამისად, ამ გადაწყვეტილებებით უნდა მოხდეს მათი ანულირება.
10) სინჯების ანალიზების მეთოდების ამ გადაწყვეტილებების დებულებებთან მისადაგების მიზნით, საჭირო გარდამავალი პერიოდის განსაზღვრა.
11) წინამდებარე გადაწყვეტილებით განსაზღვრული ზომები შეესაბამება „კვებითი ჯაჭვისა და ცხოველთა ჯანდაცვის მუდმივმოქმედი კომიტეტის“ მოსაზრებას.
მიიღო წინამდებარე გადაწყვეტილება:
მუხლი 1
საგანი და მოქმედების სფერო
მოცემული გადაწყვეტილება განსაზღვრავს ნორმებს იმ ანალიტიკურ მეთოდებთან დაკავშირებით, რომლებიც გამოიყენება დირექტივის 96/23/EC მე-15(1) მუხლის მეორე წინადადების შესაბამისად აღებული სინჯების ტესტირებისათვის და ადგენს საერთო კრიტერიუმებს განსაზღვრული ლაბორატორიების მიერ სინჯებზე ჩატარებული ანალიზების შედეგების ინტერპრეტაციისათვის.
მოცემული გადაწყვეტილების მოქმედება არ ვრცელდება იმ ნივთიერებებზე, რომლებთან დაკავშირებითაც გაერთიანების სხვა კანონმდებლობით განსაზღვრულია უფრო სპეციფიური ნორმები.
მუხლი 2
განმარტებები
ამ გადაწყვეტილების მიზნებისათვის მოქმედებს დირექტივასა 96/23/EC და დანართში მოცემული განსაზღვრებები.
მუხლი 3
ანალიტიკური მეთოდები
წევრი სახელმწიფოები უზრუნველყოფენ, რომ დირექტივის 96/23/EC შესაბამისად აღებული სინჯების ანალიზი ჩატარდება იმ მეთოდების გამოყენებით, რომლებიც:
a) დოკუმენტალურად არის წარმოდგენილი ტესტირების ინსტრუქციაში, სასურველია ISO 78-2(6) მიხედვით;
b) შეესაბამება ამ გადაწყვეტილების დანართის მე-2 ნაწილს;
c) დამტკიცებულ იქნა დანართის მე-3 ნაწილში აღწერილი პროცედურების შესაბამისად;
d) შეესაბამება განხორციელების მინიმალური მოთხოვნის ლიმიტებს (MRPL), რომლებიც განისაზღვრება მე-4 მუხლით.
მუხლი 4
მინიმალური აუცილებელი სამუშაო ზღვრები
წინამდებარე გადაწყვეტილება უნდა გადაისინჯოს, რათა მოხდეს მინიმალური აუცილებელი სამუშაო ზღვრების (MRPL) განსაზღვრა იმ ანალიზის მეთოდებისთვის, რომლებიც უნდა იქნეს გამოყენებული იმ ნივთიერებებთან მიმართებაში, რომელთათვისაც არ არის დაწესებული რაიმე ზღვარი.
მუხლი 5
ხარისხის კონტროლი
წევრი სახელმწიფოები უზრუნველყოფენ დირექტივის 96/23/EC შესაბამისად აღებული სინჯების ანალიზის შედეგების ხარისხს, განსაკუთრებით კი მონიტორინგის ტესტით მიღებულ შედეგების და/ან მარკირების შედეგების ხარისხს ISO 17025(1)-ის მე-5.9 თავის შესაბამისად.
მუხლი 6
შედეგების ინტერპრეტაცია
1. ანალიზის შედეგები უნდა ჩაითვალოს შეუსაბამოდ, თუ გადაჭარბებულია საანალიზო კომპონენტისათვის დამადასტურებელი მეთოდის ზღვრული მნიშვნელობა.
2. თუ ნივთიერებისათვის დადგენილია დასაშვები ზღვარი, მაშინ ზღვრული მნიშვნელობა წარმოადგენს კონცენტრაციას, რომლის ზემოთაც, (1 – α) სტატისტიკური სიზუსტით, შეიძლება ჩაითვალოს, რომ დასაშვები ზღვარი გადაჭარბებულია.
3. თუ ნივთიერებისათვის არ არის დადგენილი დასაშვები ზღვარი, მაშინ ზღვრული მნიშვნელობა წარმოადგენს კონცენტრაციის უმცირეს დონეს, რომლის დროსაც, (1 – α) სტატისტიკური სიზუსტით, მეთოდით შესაძლებელია საანალიზო კომპონენტის არსებობის გამოვლენა.
4. დირექტივის 96/23/EC I დანართის A ჯგუფში მითითებული ნივთიერებებისთვის ცდომილება იქნება 1% ან უფრო დაბალი. ყველა სხვა ნივთიერებებისათვის α ცდომილება იქნება 5% ან უფრო დაბალი.
მუხლი 7
გაუქმება
გაუქმდეს გადაწყვეტილებები № 90/515/EEC, 93/256/EEC და 93/257/EEC.
მუხლი 8
გარდამავალი დებულებები
დირექტივის 96/23/EC I დანართის ჯგუფში მითითებული ნივთიერებების სინჯების ანალიზის მეთოდები, რომლებიც აკმაყოფილებს გადაწყვეტილებებით 90/515/EEC, 93/256/EEC და 93/257/EEC განსაზღვრულ კრიტერიუმებს, შესაძლოა გამოყენებულ იქნეს ამ გადაწყვეტილებების ძალაში შესვლიდან ორ წლამდე პერიოდში. მეთოდები, რომლებიც ამჟამად გამოიყენება დირექტივის 96/23/EC I დანართის ჯგუფში მითითებული ნივთიერებებისათვის, შესაბამისობაში უნდა იყოს ამ გადაწყვეტილებასთან მისი ძალაში შესვლიდან არაუგვიანეს ხუთი წლის განმავლობაში.
მუხლი 9
ძალაში შესვლის თარიღი
წინამდებარე გადაწყვეტილება ძალაში შედის 2002 წლის 1 სექტემბრიდან.
მუხლი 10
ადრესატები
წინამდებარე გადაწყვეტილება ეხება წევრ სახელმწიფოებს.
შედგენილია ბრიუსელში, 2002 წლის 12 აგვისტოს.
კომისიის სახელით
დევიდ ბირნე
კომისიის წევრი
დანართი
სამუშაო კრიტერიუმები, სახვა მოთხოვნები და პროცედურები ანალიზის მეთოდებთან მიმართებაში
1. განმარტებები
1.1. სისწორე – ტესტის შედეგისა და ეტალონური მნიშვნელობის თანხვედრა(2), რომელიც დგინდება ნამდვილობისა და სიზუსტის განსაზღვრით.
1.2. ალფა (α) ცდომილება – ალბათობა იმისა, რომ გამოკვლეული ნიმუში არის შესაბამისი, მიუხედავად იმისა, რომ მიღებული იქნა შეუსაბამო შედეგი (ცრუ უარყოფითი შედეგი).
1.3. საანალიზო კომპონენტი – ის ნივთიერება, რომლის აღმოჩენა, იდენტიფიცირება ან/და ოდენობის განსაზღვრაც უნდა მოხდეს, ასევე, აღნიშნული ნივთიერების ანალიზის დროს მიღებულ დერივატივები.
1.4. ბეტა (β) ცდომილება – ალბათობა იმისა, რომ გამოკვლეული ნიმუში არის ნამდვილად შესაბამისი, მიუხედავად იმისა, რომ მიღებული იქნა შესაბამისი შედეგი (ცრუ დადებითი შედეგი).
1.5. გადახრა – სხვაობა გამოკვლევის მოსალოდნელ შედეგსა და ჭეშმარიტ (ეტალონურ) მნიშვნელობას შორის (2).
1.6. დაკალიბრების სტანდარტი – ხელსაწყო, რომლის საშუალებითაც ხდება საკვლევი ნივთიერების რაოდენობის გაზომვა, ამ მნიშვნელობასა და ეტალონურ მნიშვნელობას შორის კავშირის დადგენის მიზნით.
1.7. დაშვებული ეტალონური მასალა (CRM) – მასალა, რომლსაც გააჩნია კონკრეტული საანალიზო კომპონენტი.
1.8. თანა-ქრომატოგრაფია – პროცედურა, რომლის დროსაც ქრომატოგრაფიის ჩატარებამდე ექსტრაქტი იყოფა ორ ნაწილად. პირველ ნაწილს უტარდება ქრომატოგრაფიული ანალიზი, ხოლო მეორე ნაწილს ემატება გასაზომი სტანდარტული საანალიზო კომპონენტი. შერეულ ნიმუშს ანუ მეორე ნაწილს ასევე უტარდება ქრომატოგრაფიული ანალიზი. დამატებული სტანდარტული საანალიზო კომპონენტის რაოდენობა უნდა იყოს იმდენივე, რამდენიც შეფასების მიხედვით არსებობდა ექსტრაქტში. ეს მეთოდი მიზნად ისახავს საანალიზო კომპონენტის იდენტიფიკაციას ქრომატოგრაფიული მეთოდების გამოყენებისას, განსაკუთრებით კი იმ შემთხვევაში, როდესაც ვერ ხერხდება შესაბამისი შიდა სტანდარტის გამოყენება.
1.9. ერთობლივი კვლევა – ერთი და იგივე სინჯის ერთი და იგივე ანალიზის მეთოდით რამდენიმეჯერ გამოკვლევა, მეთოდის სამუშაო მახასიათებლების (ეფექტურობის) განსაზღვრის მიზნით. კვლევა საშუალებას იძლევა გამოვლენილი იქნეს გაზომვის შემთხვევითი ცდომილებები და ლაბორატორიული გადახრები.
1.10. დამადასტურებელი მეთოდი – მეთოდი, რომელიც იძლევა სრულ ან დამატებით ინფორმაციას, რომლის მეშვეობითაც შესაძლებელი ხდება ნივთიერების ზუსტი იდენტიფიცირება და საჭიროების შემთხვევაში, მისი მნიშვნელობის (სიდიდის) დონის ზუსტი რაოდენობის დადგენა.
1.11. ზღვრული მნიშვნელობა (CCα) – ზღვარი, რომლის ფარგლებში და ფარგლების ზემოთაც α-ს ცდომილების ალბათობით შეიძლება დადგინდეს, რომ ნიმუში შეუსაბამოა.
1.12. გამოვლენის შესაძლებლობა (CCβ) – ნივთიერების უმცირესი რაოდენობა, რომლის გამოვლენა, იდენტიფიცირება ან/და გაზომვა ნიმუშში შესაძლებელია ბეტა (β) ცდომილებით. იმ ნივთიერების შემთხვევაში, რომლისთვისაც არ არის დადგენილი დასაშვები ზღვარი, გამოვლენის შესაძლებლობას წარმოადგენს ის უმცირესი კონცენტრაცია, რომლის დროსაც მეთოდით შესაძლებელია რეალურად დაბინძურებული ნიმუშების აღმოჩენა (1-β)-ის ტოლი სტატისტიკური განსაზღვრულობით. იმ ნივთიერების შემთხვევაში, რომლისთვისაც დადგენილია დასაშვები ზღვარი, გამოვლენის შესაძლებლობას წარმოადგენს ის კონცენტრაცია, რომლის დროსაც მეთოდით შესაძლებელია დასაშვები ზღვარის კონცენტრაციის (1-β)-ის ტოლი სტატისტიკური განსაზღვრულობით.
1.13. გამდიდრებული მასალის ნიმუში (სინჯი) – განსასაზღვრი საანალიზო კომპონენტის ცნობილი რაოდენობით გამდიდრებული ნიმუში (სინჯი).
1.14. ლაბორატორიათაშორისი გამოკვლევა (შედარება) – ერთი და იმავე ნიმუშის ლაბორატორიული გამოკვლევის ორგანიზება და განხორციელება ორი ან მეტი ლაბორატორიის მიერ გამოკვლევისათვის წინასწარ განსაზღვრული პირობების შესაბამისად. მიზნების შესაბამისად, გამოკვლევა შესაძლოა კლასიფიცირებული იქნეს როგორც ერთობლივი გამოკვლევა ან საკვალიფიკაციო გამოკვლევა.
1.15. შიდა სტანდარტი (IS) – ნივთიერება, რომელსაც არ შეიცავს გამოსაკვლევი ნიმუში რომლის ფიზიკურ-ქიმიური თვისებები საანალიზო კომპონენტის თვისებების ანალოგიურია და რომელიც კვლევის დროს ემატება ყოველ ნიმუშს და ყოველ დაკალიბრების სტანდარტს.
1.16. ლაბორატორიული ნიმუში (სინჯი)– კონტროლის ან გამოკვლევის მიზნით ლაბორატორიაში გასაგზავნად გამზადებულ ნიმუში.
1.17. მნიშვნელობის (სიდიდის) დონე – ნიმუშში ნივთიერების ან საანალიზო კომპონენტის კონცენტრაცია, რომელიც საკმარისია კანონმდებლობით განსაზღვრულ მოთხოვნებთან შესაბამისობის დასადგენად.
1.18. მინიმალური აუცილებელი სამუშაო ზღვარი (MRPL) – ნიმუშში საანალიზო კომპონენტის მინიმალურ შემცველობა, რომელიც, როგორც მინიმუმ, უნდა იქნეს გამოვლენილი და დადასტურებული. აღნიშნული განკუთვნილია იმ ნივთიერებების გამოკვლევის მეთოდების ჰარმონიზაციისათვის, რომელთა მიმართაც არ არის დადგენილი დასაშვები ზღვარი.
1.19. სამუშაო მახასიათებლები – ფუნქციონალური ხარისხი, რომელიც შესაძლოა განკუთვნილი იყოს ანალიზის მეთოდისთვის. აღნიშნული შესაძლოა წარმოადგენდეს, მაგალითად, სპეციფიკურობას, სისწორეს, სანდოობას, სიზუსტეს, განმეორებადობას, შედეგების დაახლოებას, აღდგენას, გამოვლენის შესაძლებლობას და მიახლოებითობას.
1.20. სამუშაო კრიტერიუმები – სამუშაო მახასიათებლების მოთხოვნები, რომელთა მიხედვითაც შესაძლებელია დადგინდეს ანალიზის მეთოდის გამოყენების შესაბამისობა დასახულ მიზანთან და რომელიც იძლევა სანდო შედეგს.
1.21. დასაშვები ზღვარი – ნივთიერების (სუბსტანციის) გაერთიანების კანონმდებლობით განსაზღვრული ნარჩენების მაქსიმალურ ზღვარი, მაქსიმალური მნიშვნელობა ან სხვა მაქსიმალური სიდიდე.
1.22. სიზუსტე – წინასწარ განსაზღვრული პირობებით დამოუკიდებელი გამოკვლევებით მიღებული შედეგების შესაბამისობა. სიზუსტის ზომა, როგორც წესი, გამოიხატება უზუსტობით და გამოითვლება, როგორც გამოკვლევის შედეგის სტანდარტული გადახრა. ნაკლები სიზუსტე გამოითვლება უდიდესი სტანდარტული გადახრის საშუალებით (2).
1.23. საკვალიფიკაციო გამოკვლევა – ერთი და იმავე ნიმუშ(ებ)ის ანალიზი, რაც ლაბორატორიებს მეთოდების შერჩევის საშუალებას აძლევს იმ პირობით, რომ აღნიშნული მეთოდები გამოიყენება ჩვეულ პირობებში. გამოკვლევა უნდა განხორციელდეს ISO 43-1(3) და 43-2 (4)-ის შესაბამისად და შესაძლოა გამოყენებული იქნეს მეთოდების იდენტურობის (მსგავსების) შეფასებისათვის.
1.24. თვისობრივი მეთოდი – ანალიზის მეთოდი, რომელიც ახდენს ნივთიერების იდენტიფიცირებას მისი ქიმიური, ბიოლოგიური ან ფიზიკური თვისებების საფუძველზე.
1.25. რაოდენობრივი მეთოდი – ანალიზის მეთოდი, რომელიც განსაზღვრავს ნივთიერების რაოდენობას ან მასურ წილს, იმისათვის, რომ აღნიშნულის გამოსახვა შესაძლებელი იყოს შესაბამისი ერთეულის რიცხვითი მნიშვნელობით.
1.26. სუფთა რეაქტივის განსაზღვრა – სრული ანალიზი, რომლის დროსაც არ გამოიყენება ნიმუში, ან ნიმუშის ნაცვლად გამოიყენება შესაბამისი გამხსნელის ეკვივალენტური რაოდენობა.
1.27. აღდგენა – ნივთიერების ჭეშმარიტი კონცენტრაციის პროცენტული შემცველობა, რომელიც მიიღება ანალიზის დროს. აღნიშნული განისაზღვრება ვალიდაციის პროცესში იმ შემთხვევაში, თუ ატესტირებული (დაშვებული) ეტალონური მასალა (CRM) არ არის ხელმისაწვდომი.
1.28. ეტალონური მასალა – ნივთიერება, რომლის ერთი ან რამდენიმე თვისება დადასტურებული იქნა ვალიდური მეთოდით ისე, რომ ამ მასალის გამოყენება შესაძლებელია ხელსაწყოს დაკალიბრების ან გაზომვის მეთოდის შემოწმების მიზნით.
1.29. განმეორებადობა – სიზუსტე განმეორებადობის პირობებში (2).
1.30. განმეორებადობის პირობები – პირობები, როდესაც დამოუკიდებელი გამოკვლევის შედეგები მიღება იდენტური საკვლევი მასალის ანალიზის დროს, ერთი და იგივე მეთოდის გამოყენებით, რომელიც ტარდება ერთი და იგივე ლაბორატორიაში, ერთი და იმავე სპეციალისტის (ოპერატორის) მიერ, ერთი და იგივე აპარატურის გამოყენებით (2).
1.31. აღწარმოებადობა - სიზუსტე აღწარმოებადობის პირობებში (2).
1.32. აღწარმოებადობის პირობები – პირობები, როდესაც გამოკვლევის შედეგები მიიღება იდენტური საკვლევი მასალის ანალიზისას, ერთი და იგივე მეთოდის გამოყენებით, რომელიც ტარდება სხვადასხვა ლაბორატორიაში, სხვადასხვა სპეციალისტის (ოპერატორის) მიერ, სხვადასხვა აპარატურის გამოყენებით (2)(4).
1.33. მდგრადობა – ექსპერიმენტული პირობების შეცვლისას ანალიზის მეთოდის მგრძნობელობა, რომელიც შესაძლოა გამოიხატოს ნიმუშის მასალების, საანალიზო კომპონენტების, შენახვისა და გარემო პირობების ან/და ნიმუშის მომზადების პირობების ნუსხის სახით, რომლის დროსაც შესაძლებელია გამოყენებული იქნეს მეთოდი წარმოდგენილი სახით ან მცირედი შესწორებებით. ყველა ექსპერიმენტულ პირობებში, რომლებიც პრაქტიკაში გამოყენებისას შეიძლება გახდეს ცვალებადი, (მაგ.: რეაქტივების მდგრადობა, ნიმუშის შემადგენლობა, pH, ტემპერატურა) აუცილებელია მითითებულ იქნეს ნებისმიერი ცვლილება, რომელმაც შესაძლოა ზეგავლენა მოახდინოს ანალიზის შედეგზე.
1.34. სუფთა ნიმუშის განსაზღვრა – სრული ანალიზი, რომელიც გამოიყენება ნიმუშის იმ ნაწილის გამოკვლევისათვის, რომელშიც არ არის საანალიზო კომპონენტი.
1.35. სკრინინგის მეთოდი – მეთოდი, რომელიც გამოიყენება ნივთიერების ან ნივთიერებათა კლასის დადგენისათვის მისი მნიშვნელობის (სიდიდის) დონეზე. ამ მეთოდს გააჩნია ნიმუშის მაღალი გამტარუნარიანობის შესაძლებლობა და მისი გამოყენება ხდება დიდი ოდენობის პოტენციურად შეუსაბამო, უარყოფითი ნიმუშების გადარჩევისათვის. აღნიშნული მეთოდი სპეციალურად არის შემუშავებული ცრუ დადებითი შედეგების თავიდან აცილების მიზნით.
1.36. ერთი ლაბორატორიის გამოკვლევა (შიდა ვალიდაცია) – ერთი ლაბორატორიის მიერ ჩატარებული ანალიზური გამოკვლევა ერთი და იმავე, ან განსხვავებული საკვლევი მასალების, ერთი მეთოდის გამოყენებით, ჩატარებულ გამოკვლევას დასაბუთებულ გრძელვადიან ინტერვალებში.
1.37. თავისებურება (სპეციფიკურობა) – მეთოდის შესაძლებლობა, ანალიზის დროს განასხვავოს საანალიზო კომპონენტი სხვა ნივთიერებისაგან. აღნიშნული მახასიათებელი წარმოადგენს გაზომვის მეთოდის ფუნქციას, მაგრამ შესაძლებელია შეიცვლოს ნივთიერების კლასის ან მატრიცის შესაბამისად.
1.38. სტანდარტული ნივთიერების დამატება – პროცედურა, რომლის დროსაც საკვლევი ნიმუში იყოფა ორ (ან მეტ) საკვლევ ნაწილად. ერთი ნაწილს პირდაპირ უტარდება ანალიზი, ხოლო დანარჩენ ნაწილ(ებ)ს ანალიზამდე ემატება საანალიზო კომპონენტის ცნობილი რაოდენობა. დამატებული სტანდარტული საანალიზო კომპონენტის რაოდენობა 2-5-ჯერ უნდა აღემატებოდეს ნიმუშში საკვლევი საანალიზო კომპონენტის რაოდენობას. პროცედურა შემუშავებულია ნიმუშში საანალიზო კომპონენტის განსაზღვრისათვის, ანალიზის აღდგენის გათვალისწინებით.
1.39. სტანდარტული საანალიზო კომპონენტი – ანალიზის დროს გამოსაყენებელი ცნობილი და განსაზღვრული შემადგენლობისა და სისუფთავის მქონე ეტალონურ ნივთიერება.
1.40. ნივთიერება – კონკრეტული ან განსაზღვრული ქიმიური შემადგენლობის მასალა და მისი მეტაბოლიტები.
1.41. საკვლევი (სატესტო) ნაწილი – ლაბორატორიული გამოკვლევისათვის განკუთვნილი, საკვლევი ნიმუშიდან აღებული მასალის გარკვეული რაოდენობა.
1.42. საკვლევი სინჯი – სინჯი, რომელიც მიიღება ლაბორატორიული გამოკვლევისათვის განკუთვნილი ნიმუშისგან (სინჯისაგან) და რომლისგანაც აღებული იქნება საკვლევი ნაწილები.
1.43. სანდოობა – დიდი რაოდენობის კვლევის შედეგებიდან მიღებული საშუალო სიდიდესა და აღიარებულ ეტალონურ სიდიდეს შორის შესაბამისობის სიზუსტე. სანდოობა ჩვეულებრივ გამოიხატება, როგორც გადახრა (2).
1.44. ერთეულები – ISO 31 (20) და ევროკავშირის 71/354/ EC დირექტივით განსაზღვრული ერთეულები.
1.45. ვალიდაცია – გამოკვლევითა და ეფექტური მტკიცებულებების წარდგენით იმის დადასტურება, რომ სრულდება კონკრეტული მიზნობრივი გამოყენებისთვის განსაზღვრული ყველა მოთხოვნა (1).
1.46. შიდალაბორატორიული აღწარმოებადობა – შედეგის სიზუსტის განმეორებადობა ერთი და იმავე ლაბორატორიაში, წინასწარ დადგენილ პირობებში (მაგ.: მეთოდი, საკვლევი მასალა, სპეციალისტი, გარემო), დროის დასაბუთებულ ხანგრძლივ ინტერვალებში.
2. სამუშაო კრიტერიუმები და სხვა მოთხოვნები ანალიზის მეთოდებისადმი
ანალიზის მეთოდები, ან ანალიზის მეთოდების ერთობლიობა, გარდა ქვემოთ აღწერილი მეთოდებისა, შესაძლებელია გამოყენებული იქნეს მხოლოდ სკრინინგის ან დამადასტურებელი მიზნისათვის, თუ დადასტურებული იქნება, რომ ისინი აკმაყოფილებენ წინამდებარე გადაწყვეტილებით განსაზღვრულ მოთხოვნებს.
2.1. ზოგადი მოთხოვნები
2.1.1 ნიმუშის მომზადება
ნიმუშების აღება, დამუშავება და გადამუშავება უნდა მოხდეს ისე, რომ მაქსიმალური ალბათობით შესაძლებელი იქნეს ნივთიერების აღმოჩენა. ნიმუშის მომზადება უნდა გამორიცხავდეს საანალიზო კომპონენტის შემთხვევით დაბინძურების, ან დაკარგვის შესაძლებლობას.
2.1.2 კვლევების განხორციელება
2.1.2.1. აღდგენა
თითოეული პარტიისათვის, ნიმუშების ანალიზის დროს, უნდა განისაზღვროს აღდგენა, თუ გამოიყენება აღდგენის მუდმივი შესწორების კოეფიციენტი. თუ აღდგენა ხდება გარკვეულ ზღვრებში, მაშინ შეიძლება გამოყენებულ იქნეს მუდმივი შესწორების კოეფიციენტი. სხვა შემთხვევაში, გამოყენებული უნდა იქნეს აღნიშნული კონკრეტული პარტიისთვის განსაზღვრული აღდგენის ფაქტორი, გარდა იმ შემთხვევისა, როდესაც ანალიზის დროს გამოიყენება ნიმუშში საანალიზო კომპონენტის სპეციფიკური აღმდგენი ფაქტორი. ამ შემთხვევაში, ნიმუშში საანალიზო კომპონენტის რაოდენობრივი განსაზღვრისათვის გამოყენებული უნდა იქნეს სტანდარტის დამატების ან შიდა სტანდარტის დამატების პროცედურა.
2.1.2.2. სპეციფიკურობა
ექსპერიმენტულ პირობებში, ანალიზის ჩატარების მეთოდი საანალიზო კომპონენტის და სხვა ნივთიერებების ერთმანეთისგან განსხვავების შესაძლებლობას უნდა იძლეოდეს. შეფასება აუცილებლად უნდა მოხდეს რამდენადაც ეს შესაძლებელია. გაზომვის აღწერილი მეთოდის გამოყენებისას, გათვალისწინებული უნდა იქნეს ნივთიერების ნებისმიერი სავარაუდო ინტერფერენციის თავიდან აცილების სტრატეგია, მაგ.: საკვლევი ნარჩენების ჰომოლოგები, ანალოგები, მეტაბოლიტები. მნიშვნელოვანია ინტერფერენციის შესწავლა, რომელიც შესაძლებელია წარმოიქმნას მატრიცული კომპონენტებიდან.
2.2. სკრინინგის მეთოდი
სკრინინგის მიზნებისათვის, დირექტივის 96/23/EC შესაბამისად, გამოყენებული უნდა იქნეს მხოლოდ ანალიზის ის მეთოდები, რომლებთან მიმართებაშიც შესაძლებელია დოკუმენტურად იქნეს დასაბუთებული, რომ ისინი ვალიდურია და აქვთ მცდარი შესაბამისობის კოეფიციენტი, რომელიც ნაკლებია 5 %-ზე (β-ცდომილება). მოსალოდნელი შეუსაბამო შედეგის შემთხვევაში, აღნიშნული შედეგი უნდა დადასტურდეს დამადასტურებელი მეთოდის საშუალებით.
2.3. ორგანული ნარჩენებისა და დამაბინძურებლების აღმოჩენის დამადასტურებელი მეთოდები
ორგანული ნარჩენებისა ან დამაბინძურებლების აღმოჩენის დამადასტურებელი მეთოდები უნდა იძლეოდნენ საანალიზო კომპონენტის ქიმიური სტრუქტურის ამსახველ ინფორმაციას. შესაბამისად, მხოლოდ ქრომატოგრაფიულ ანალიზზე დაფუძნებული მეთოდების გამოყენება დამოუკიდებლად, სპექტრომეტრული მეთოდის გარეშე, არ შეიძლება ჩაითვალოს დამადასტურებელ მეთოდად. თუ რომელიმე ცალკეული მეთოდი არასაკმარისად სპეციფიკურია, მაშინ სასურველი სპეციფიკურობის მიღწევა შესაძლებელია ანალიზური პროცედურებით, რომლებიც მოიცავს გაწმენდის, ქრომატოგრაფიული დანაწილების და სპექტრომეტრული გამოვლენის მეთოდების ერთობლიობას.
ორგანული ნარჩენების ან დამაბინძურებლების იდენტიფიკაციისათვის დადგენილია შემდეგი მეთოდები ან მეთოდთა ერთობლიობა ქვემოთ მითითებული ნივთიერებების შემთხვევაში:
ცხრილი №1
ორგანული ნარჩენებისა და დამაბინძურებლების აღმოჩენის დამადასტურებელი მეთოდები
|
გაზომვის მეთოდი |
ნივთიერებები დანართი 1 96/23/EC |
შეზღუდვები |
|
LC ან GC მას-სპექტტრომეტრული დეტექციით
LC ან GC ინფრაწითელი სპექტრომეტრული დეტექციით
LC – სრული სკანირება DAD
LC – ფლუორესცენცია
2-D TLC – სრული სკანირება UV/VIS
GC – ელექტრონის დაჭერით დეტექცია
LC – იმუნოგრამა
LC – UV/VIS (ერთი-ტალღის-სიგრძის) |
ჯგუფები A და B
ჯგუფები A და B
ჯგუფი B
ჯგუფი B
ჯგუფი B
ჯგუფი B
ჯგუფი B
ჯგუფი B |
მხოლოდ იმ შემთხვევაში, თუ მოჰყვება on-line ან off-line ქრომოტოგრაფიულ განცალკევებას.
მხოლოდ იმ შემთხვევაში, თუ გამოიყენება სრული სკანირების მეთოდი ან, როგორც მინიმუმ, 3 (ჯგუფი B) or 4 (ჯგუფი A) საიდენტიფიკაციო წერტილები იმ მეთოდებისთვის, რომელთა გამოყენებისას არ ხდება სრული მასური სპექტრის ჩაწერა.
დაკმაყოფილებული უნდა იქნეს აბსორბციასთან დაკავშირებული კონკრეტული მოთხოვნები IR სპეტრომეტრიაში.
დაკმაყოფილებული უნდა იქნეს აბსორბციასთან დაკავშირებული კონკრეტული მოთხოვნები UV სპექტრომეტრიაში.
მხოლოდ იმ მოლეკულებისთვის, რომლებიც ავლენენ ბუნებრივ ფლუორესცენციას და იმ მოლეკულებისთვის, რომლებიც ავლენენ ფლუორესცენციას ტრანსფორმაციის ან დერივატიზაციის შემდგომ.
ორგანზომილებიანი HPTLC და თანაქრომატოგრაფია სავალდებულოა მხოლოდ იმ შემთხვევაში, თუ გამოიყენება ორი სხვადასხვა პოლარულობის სვეტი.
მხოლოდ იმ შემთხვევაში, თუ სულ მცირე ორი სხვადასხვა ქრომატოგრაფიული სისტემა ან მეორე, დამოუკიდებელი დეტექციის მეთოდი არის გამოყენებული.
მხოლოდ იმ შემთხვევაში, თუ სულ მცირე ორი სხვადასხვა ქრომატოგრაფიული სისტემა ან მეორე, დამოუკიდებელი დეტექციის მეთოდი არის გამოყენებული. |
2.3.1. საერთო სამუშაო კრიტერიუმები და მოთხოვნები
დამადასტურებელი მეთოდები უნდა იძლეოდეს საანალიზო კომპონენტის ქიმიური სტრუქტურის ამსახველ ინფორმაციას. იმ შემთხვევაში, როდესაც ერთზე მეტი ნაერთი იძლევა ერთი და იმავე რეაქციას, აღნიშნული მეთოდის გამოყენებით ვერ განხორციელდება ამ ნაერთების ერთმანეთისგან განსხვავება. შესაბამისად, მხოლოდ ქრომატოგრაფიულ ანალიზზე დაფუძნებული მეთოდების გამოყენება დამოუკიდებლად, სპექტრომეტრული მეთოდის გარეშე, არ შეიძლება ჩაითვალოს დამადასტურებელ მეთოდად.
მეთოდში გამოყენების შემთხვევაში, შესაბამისი შიდა სტანდარტი უნდა დაემატოს ნიმუშის საკვლევ ნაწილს ექსტრაქციის პროცედურების დაწყებამდე. ხელმისაწვდომობის შესაბამისად, გამოყენებული უნდა იქნეს საანალიზო კომპონენტების სტაბილური იზოტოპებით ნიშანდებული ფორმები, რომლებიც განსაკუთრებით შესაფერისია მასობრივი სპექტომეტრიისთვის, ან ნაერთები, რომლებიც სტრუქტურულად დაკავშირებულია საანალიზო კომპონენტთან
შესაფერისი შიდა სტანდარტის გამოყენების შეუძლებლობის შემთხვევაში, საანალიზო კომპონენტის იდენტიფიკაციის დადასტურება უნდა განხორციელდეს თანაქრომატოგრაფიით. ამ შემთხვევაში მიიღება მხოლოდ ერთი პიკი, სადაც გაზრდილი პიკის სიმაღლე (ან ფართობი) დამატებული საანალიზო კომპონენტის ეკვივალენტურია. გაზის ქრომატოგრაფიის (GC) ან სითხის ქრომატოგრაფიის (LC) გამოყენებისას, პიკის სიგანე, პიკის მაქსიმალური სიმაღლის ნახევარზე, უნდა იყოს თავდაპირველი სიგანის 90-110% დიაპაზონის ფარგლებში, ხოლო შეკავების დრო უნდა იყოს ერთნაირი 5%-ის ცდომილების ფარგლებში. თხელფენოვანი ქრომატოგრაფიის (TLC) მეთოდებისათვის, მკაფიო ხდება მხოლოდ ლაქა, რომელიც სავარაუდოდ წარმოიქმნა საანალიზო კომპონენტიდან; არ ხდება ახალი ლაქის წარმოქმნა და ვიზუალურად გარეგანი სახის ცვლილება.
ეტალონური ან გამდიდრებული მასალა, რომელიც შეიცავს საანალიზო კომპონენტის ცნობილ რაოდენობას, დასაშვები ზღვარის ან ზღვრული მნიშვნელობის (შეუსაბამო საკონტროლო ნიმუშის) ფარგლებში, ასევე შესაბამისი საკონტროლო მასალა და სუფთა რეაქტივები გამოყენებული უნდა იქნეს ანალიზის მსვლელობის პროცედურების განმავლობაში, საანალიზო ნიმუშების თითოეულ პარტიასთან ერთად. საანალიზო ხელსაწყოში ექსტრაქტების შეყვანა ხდება შემდეგი თანმიმდევრობით: სუფთა რეაქტივი, შესაბამისი საკონტროლო ნიმუში, ნიმუშ(ებ)ი, რომელთა დადასტურებაც აუცილებელია, ისევ შესაბამისი საკონტროლო ნიმუში და ბოლოს, შეუსაბამო საკონტროლო ნიმუში. ამ თანმიმდევრობის ნებისმიერი ცვლილება უნდა იქნეს დასაბუთებული.
2.3.2. დამატებითი სამუშაო კრიტერიუმები და სხვა მოთხოვნები ანალიზის რაოდენობრივი მეთოდებისადმი
2.3.2.1. რაოდენობრივი მეთოდების სანდოობა
დაშვებული ეტალონური მასალის განმეორებითი ანალიზის შემთხვევაში, დაშვებული მნიშვნელობებიდან აღდგენის მასური წილის საშუალო მნიშვნელობის გადახრის ექსპერიმენტულად განსაზღვრული, შესწორებული, რეკომენდებული დიაპაზონი განისაზღვრება შემდეგნაირად:
ცხრილი №2
რაოდენობრივი მეთოდების მინიმალური სანდოობა
|
მასური წილი |
დიაპაზონი |
|
≤ 1 µg/kg |
– 50 % to + 20 % |
|
> 1 µg/kg to 10 µg/kg |
– 30 % to + 10 % |
|
≥ 10 µg/kg |
– 20 % t o + 10 % |
თუ არ არის ხელმისაწვდომი ასეთი დაშვებული ეტალონური მასალა (CRM), დასაშვებია გაზომვის სანდოობა შეფასებულ იქნეს სუფთა მატრიცაზე საანალიზო კომპონენტის ცნობილი რაოდენობების დამატებების აღდგენით; აღდგენის საშუალო მნიშვნელობით შესწორებული მონაცემები გამოიყენება მხოლოდ იმ შემთხვევაში, თუ ისინი შეესაბამება ცხრილი 2-ით განსაზღვრულ დიაპაზონის ფარგლებს.
2.3.2.2. რაოდენობრივი მეთოდების სიზუსტე
ეტალონური ან გამდიდრებული მასალის განმეორებითი ანალიზის დროს, ლაბორატორიათშორისი ვარიაციის კოეფიციენტი (CV), აღწარმოებადობის პირობებში, არ უნდა აღემატებოდეს ჰორვიცის (Horwitz) განტოლებით გამოთვლილ დონეს, განტოლება:
CV = 2(1 – 0,5 log C)
სადაც C – მასური წილია, რომელიც გამოსახულია ხარისხით (ხარისხის მაჩვენებლით) და ტოლია 10-ის (მაგ. 1მგ/გ = 10-3) მაგალითები მოცემულია ცხრილ №3-ში
ცხრილი №3
ვარიაციის კოეფიციენტების (CV) აღწარმოებადობის მაგალითები რაოდენობრივი მეთოდებისათვის საანალიზო კომპონენტის მასური წილის დიაპაზონის მიხედვით
|
მასური წილი |
აღწარმოებადობა CV(%) |
|
1 µg/kg |
(*) |
|
10 µg/kg |
(*) |
|
100 µg/kg |
23 |
|
1 000 µg/kg (1 mg/kg |
16 |
|
(*)100 µg/kg-ზე ნაკლები მასური წილების შემთხვევაში Horwitz-ის განტოლების გამოყენება გავძლევს მიუღებელ მაღალ მნიშვნელობებს. ამიტომ, ვარიაციის კოეფიციენტები 100 µg/kg-ზე ნაკლები კონცენტრაციებისთვის უნდა იყოს მაქსიმალურად დაბალი. |
|
განმეორებადობის პირობებში ჩატარებული ანალიზისათვის, ლაბორატორიათშორისი ვარიაციის კოეფიციენტი (CV), როგორც წესი, მდებარეობს ზემოთ მითითებულ მნიშვნელობათა ნახევარსა და ორ მესამედს შორის. აღწარმოებადობის პირობებში ჩატარებული შიდალაბორატორიული ანალიზისას შიდალაბორატორიული ვარიაციის კოეფიციენტი არ უნდა აღემატებოდეს აღწარმოებადობის ვარიაციის კოეფიციენტს (CV).
იმ ნივთიერებ(ებ)ის შემთხვევაში, რომელთა დასაშვები ზღვარი დადგენილია, მეთოდი აღწევს შიდალაბორატორიულ აღწარმოებადობას, რომელიც არ აღემატება შესაბამის აღწარმოებადობის ვარიაციის კოეფიციენტს (CV), 0,5 × დასაშვები ზღვარი კონცენტრაციის პირობებში.
2.3.3. სამუშაო კრიტერიუმები და სხვა მოთხოვნები მასის სპექტრომეტრული გამოვლენისადმი
მას-სპექტომეტრული მეთოდების დამადასტურებელი მეთოდის სახით გამოყენება დასაშვებია მხოლოდ on-line ან off-line ქრომატოგრაფიული დაყოფის შემდეგ.
2.3.3.1. ქრომატოგრაფიული დაყოფა
GC-MS – პროცედურებისთვის გაზის ქრომატოგრაფიული დაყოფა ხორციელდება კაპილარული სვეტების გამოყენებით. LC-MS პროცედურებისთვის ქრომატოგრაფიული დაყოფა უნდა განხორციელდეს შესაბამისი LC სვეტების გამოყენებით. ნებისმიერ შემთხვევაში, საანალიზო კომპონენტის შეკავების მისაღები მინიმალური დრო ორჯერ აღემატება იმ შეკავების დროს, რომელიც შეესაბამება სვეტის თავისუფალ მოცულობას. საკვლევ ნაწილში საანალიზო კომპონენტის შეკავების დრო (ან შეკავების ფარდობითი დრო) უნდა შეესაბამებოდეს დაკალიბრების სტანდარტის შეკავების დროს შეკავების განსაზღვრული ინტერვალის ფარგლებში. შეკავების დროის ინტერვალი ქრომატოგრაფიული სისტემის გარჩევადობის უნარის პროპორციული უნდა იყოს. საანალიზო კომპონენტის ქრომატოგრაფიული შეკავების დროის შეფარდება შიდა სტანდარტის შეკავების დროსთან, ანუ საანალიზო კომპონენტის შეკავების ფარდობითი დრო, შეესაბამება საკალიბრო ხსნარის შეკავების დროს დაშვებული გადახრებით, რომელიც GC-თვის შეადგენს ± 0,5 %-ს, ხოლო LС- თვის - ± 2,5 %-ს.
2.3.3.2. მას-სპექტომეტრული გამოვლენა
მას-სპექტომეტრული გამოვლენა უნდა განხორციელდეს ისეთი MS – მეთოდებით, როგორიცაა სრული მას-სპექტრის რეგისტრაცია (სრული სკანირება) ან იონების შერჩევითი მონიტორინგი (SIM), აგრეთვე ისეთი MS-MSn მეთოდებით, როგორიცაა რეაქციის შერჩევითი მონიტორინგი (SRM), ან სხვა შესაბამისი MS ან MS-MSn მეთოდებით, იონიზაციის შესაბამის რეჟიმებთან ერთობლიობაში. მაღალი გარჩევადობის მას-სპექტომეტრიაში (HRMS), გარჩევადობის უნარი, როგორც წესი, მთლიანი მასური დიაპაზონისთვის 10% ველზე უნდა იყოს 10 000-ზე მეტი.
სრული სკანირება როდესაც მას-სპექტომეტრული განსაზღვრა ხორციელდება სპექტრის სრული სკანირების რეგისტრაციით, სავალდებულოა ყველა გასაზომი სადიაგნოსტიკო იონების არსებობა (მოლეკულური იონი, მოლეკულური იონის დამახასიათებელი ადუქტები, იონების დამახასიათებელი ფრაგმენტები და იზოტოპური იონები) დაკალიბრების სტანდარტის ეტალონურ სპექტრში 10 %-ზე მეტი ფარდობითი ინტენსივობით.
იონების შერჩევითი მონიტორინგი (SIM) როდესაც მას-სპექტრომეტრული განსაზღვრა ხორციელდება ფრაგმენტოგრაფით, მოლეკულური იონი სასურველია იყოს ერთ-ერთი სადიაგნოსტიკო იონი (მოლეკულური იონი, მოლეკულური იონის დამახასიათებელი ადუქტები, იონების დამახასიათებელი ფრაგმენტები და ყველა მათი იზოტოპური იონები). შერჩეული სადიაგნოსტიკო იონები არ უნდა გამომდინარეობდნენ მოლეკულის ერთი და იმავე ნაწილიდან. სიგნალ-ხმაურის თანაფარდობა ყოველი სადიაგნოსტიკო იონისათვის უნდა იყოს ≥ 3:1.
სრული სკანირება და იონების შერჩევითი მონიტორინგი (SIM): გამოვლენილი იონების ფარდობითი ინტენსივობა, რომელიც გამოხატულია, როგორც ყველაზე უფრო ინტენსიური იონის ინტენსივობის შემცველობა (პროცენტებში) ან გარდაქმნა, უნდა შეესაბამებოდეს დაკალიბრების სტანდარტის იონების ფარდობით ინტენსივობას, რომლებიც მიღებულია დაკალიბრების სტანდარტული ხსნარებიდან, ან გარკვეული დანამატების შემცველი ნიმუშებიდან, შედარებითი კონცენტრაციის პირობებში, რომელიც იზომება ერთნაირ პირობებში შემდეგი ცდომილებების პირობებში.
ცხრილი №4
მაქსიმალური დასაშვები ცდომილებები იონების ფარდობითი ინტენსივობისთვის მთელი რიგი მას-სპექტრომეტრული მეთოდების გამოყენების დროს
|
ფარდობითი ინტენსივობა (საბაზისო პიკის %) |
EI-GC-MS (ფარდობითი) |
CI-GC-MS, GC-MSn LC-MS, LC-MSn (ფარდობითი) |
|
> 50%
> 20% to 50 %
> 10 % to 20 %
≤ 10 % |
± 10%
± 15%
± 20 %
± 50 % |
± 20%
± 25%
± 30 %
± 50 % |
მას-სპექტრალური მონაცემების ინტერპრეტაცია სადიაგნოსტიკო იონების ან/და იონური წყვილების პრეკურსორის/პროდუქტის ფარდობითი ინტენსივობის განსაზღვრა ხდება სპექტრების შედარებით ან ერთეული მასის კვალის სიგნალების ინტეგრაციით. უკანა ფონის შესწორების გამოყენების შემთხვევაში, ის გამოყენებული უნდა იქნეს მთლიანი პარტიისათვის (სერიისათვის) ერთგვაროვნად (იხ. 2.3.1, მე-4 აბზაცი) და ამის შესახებ გარკვევით უნდა იქნეს მითითებული.
სრული სკანირება: როდესაც ერთეული მასის სპექტომეტრით ხდება სრული სკანირების სპექტრების რეგისტრაცია, მას თან უნდა ახლდეს არანაკლებ ოთხი იონი საბაზისო პიკის ფარდობითი ინტენსივობით ≥ 10 %. მოლეკულური იონის ჩართვა ხდება, თუ ის არსებობს ეტალონური სპექტრის ≥ 10% ფარდობითი ინტენსივობით. იონების ფარდობითი ინტენსივობისათვის არანაკლებ ოთხი იონი უნდა არსებობდეს დასაშვები ცდომილების საზღვრებში (ცხრილი №5). შესაძლებელია გამოყენებული იქნეს კომპიუტერული ბიბლიოთეკის მონაცემთა ბაზა. ამ შემთხვევაში საკვლევ ნიმუშში მას-სპექტრალური მონაცემების და საკალიბრო ხსნარის მონაცემების შეფარდება უნდა აღემატებოდეს შესაბამის კრიტიკულ ფაქტორს. ამ ფაქტორის განსაზღვრა ხდება ვალიდაციის პროცესისას თითოეული საანალიზო კომპონენტისთვის იმ სპექტრების მიხედვით, რომლებისთვისაც სრულდება ქვემოთ აღწერილი კრიტერიუმები. ხდება ასევე სპექტრში ცვლილებების შესწავლა, რომელიც შეიძლება გამოწვეული იყოს ნიმუშის მატრიცით და დეტექტორის სამუშაო მახასიათებლებით.
იონების შერჩევითი მონიტორინგი (SIM): მას-ფრაგმენტების სხვა მეთოდით განსაზღვრისას, სრული სკანირების მეთოდთან ერთად, მონაცემთა შეფასებისათვის გამოიყენება იდენტიფიკაციის წერტილების სისტემა. დირექტივის 96/23/EC I დანართის A ჯგუფში მითითებული ნივთიერებების დადასტურებისთვის საჭიროა სულ მცირე 4 იდენტიფიკაციის წერტილი. დირექტივის 96/23/EC I დანართის B ჯგუფში მითითებული ნივთიერებების დადასტურებისთვის საჭიროა სულ მცირე 3 იდენტიფიკაციის წერტილი. ქვემოთ მოცემულ ცხრილში განსაზღვრულია იდენტიფიკაციის წერტილები, რომლებიც შესაძლებელია მიღებულ იქნეს თითოეული ძირითადი მას-სპექტრომეტრული მეთოდით. თუმცა, იმისათვის რომ განისაზღვროს დადასტურებისთვის აუცილებელი იდენტიფიკაციის წერტილები, ასევე გამოსათვლელი იდენტიფიკაციის წერტილების ჯამი:
a) გაზომილი უნდა იქნეს არანაკლებ იონების ერთი თანაფარდობა; და
b) ყველა შესაბამისი გაზომილი იონების თანაფარდობა უნდა აკმაყოფილებდეს ზემოთ აღწერილ კრიტერიუმებს; და
c) იდენტიფიკაციის წერტილების მინიმალური რაოდენობის მისაღებად დასაშვებია არა უმეტეს სამი სხვადასხვა მეთოდის გაერთიანება.
ცხრილი №5
მასური ფრაგმენტების კლასებსა და მიღებულ იდენტიფიკაციის წერტილებს შორის ურთიერთობა
|
MS მეთოდი |
თითოეულ იონზე მიღებული საიდენტიფიკაციო წერტილები |
|
|
დაბალი გარჩევადობის მას-სპექტრომეტრია (LR)
LR-MSn პრეკურსორი იონი
LR-MSn გარდამავალი პროდუქტები
HRMS
HR- MSn პრეკურსორი იონი
HR-MSn გარდამავალი პროდუქტები |
1,0
1,0
1,5
2,0
2,0
2,5 |
სქოლიო:
1) თითოეული იონის ჩათვლა დასაშვებია მხოლოდ ერთხელ.
2) ის GC-MS მეთოდი, რომელიც იყენებს ელექტრონის დარტყმით იონიზაციას, ითვლება განსხვავებულად იმ GC-MS მეთოდისგან, რომელიც იყენებს ქიმიურ იონიზაციას.
3) სხვადასხვა საანალიზო კომპონენტი შეიძლება იქნეს გამოყენებული იდენტიფიკაციის წერტილების რაოდენობის გასაზრდელად მხოლოდ იმ შემთხვევაში, თუ მათი მიღება ხდება სხვადასხვა ქიმიური რეაქციებით.
4) დირექტივის 96/23/EC I დანართის A ჯგუფში მითითებული ნივთიერებების შემთხვევაში, თუ ანალიზისას გამოიყენება ერთ-ერთი შემდეგი მეთოდი: HPLC, დაწყვილებული დიოდური მატრიცის (DAD) სრული სკანირების სპექტომეტრიასთან; HPLС, დაწყვილებული ფლუორესცენტულ დეტექციასთან; HPLC, დაწყვილებული იმუნოგრამასთან; ორგანზომილებიანი TLC, დაწყვილებული სპქტრომეტრულ დეტექციასთან; შესაძლებელია მხოლოდ ერთი იდენტიფიკაციის წერტილის შეტანა, იმ პირობით, რომ სრულდება აღნიშნული მეთოდების შესაბამისი კრიტერიუმები.
5) გარდამავალი პროდუქტები მოიცავს დაშლის როგორც მეორე, ასევე მესამე თაობის პროდუქტებს.
ცხრილი №6
ცალკეული მეთოდებისა და მათი ერთობლიობისას მიღებული იდენტიფიკაციის წერტილების რაოდენობის მაგალითები
|
მეთოდ(ებ)ი |
იონების რაოდენობა |
იდენტიფიკაციის წერტილები |
|
GC-MS (EI ან CI)
GC-MS (EI და CI)
GC-MS (EI ან CI) 2 დერივატივები
LC-MS
GC-MS-MS
LC-MS-MS
GC-MS-MS
LC-MS-MS
LC-MS-MS-MS
HRMS
GC-MS and LC-MS
GC-MS and HRMS |
N
2 (EI) + 2 (CI)
2 (დერივატივი A) + 2 (დერივატივი B)
N
1 პრეკურსორი და 2 მეორე თაობის პროდუქტით
1 პრეკურსორი და 2 მეორე თაობის პროდუქტით
2 პრეკურსორი იონები, თითოეული 1 მეორე თაობის პროდუქტით
2 პრეკურსორი იონი, თითოეული 1 მეორე თაობის პროდუქტით
1 პრეკურსორი, 1 მეორე თაობის და 2 მესამე თაობის პროდუქტით
N
2 + 2
2 + 1 |
n
4
4
n
4
4
5
5
5,5
2 n
4
4 |
2.3.4. ინფრაწითელ დეტექციასთან დაწყვილებული ქრომატოგრაფიის სამუშაო კრიტერიუმები
ადეკვატური პიკი – ადეკვატური პიკი წარმოადგენს ინფრაწითელ სპექტრში დაკალიბრების სტანდარტის შთანთქმის (აბსორბციის) მაქსიმალურ მაჩვენებელს, რომელიც აკმაყოფილებს შემდეგ მოთხოვნებს.
2.3.4.1. ინფრაწითელი დეტექცია
შთანთქმის მაქსიმუმი: უნდა მოექცეს 4 000-500 სმ-1 ტალღური რიცხვის დიაპაზონში.
შთანთქმის ინტენსივობა: არ უნდა იყოს ერთერთ შემდეგ მონაცემზე ნაკლები:
(a) კუთრი მოლური შთანთქმის უნარი – 40 %, საბაზისო ხაზის პიკთან მიმართებაში; ან
(b) შთანთქმის ფარდობითი უნარი – 12,5 %, იმ პიკთან მიმართებაში, რომელსაც 4000 – 500სმ-1 დიაპაზონში აქვს ყველაზე ინტენსიური შთანთქმის უნარი,
როდესაც ორივე გამოთვლები ხორციელდება ნულოვან შთანთქმასთან მიმართებაში, და ყველაზე ინტენსიური პიკის შთანთქმის უნარის 5%-თან მიმართებაში 4 000 – 500სმ-1 დიაპაზონში, როდესაც ორივე იზომება მათივე საბაზისო ხაზის პიკთან მიმართებაში.
შენიშვნა:
მიუხედავად იმისა, რომ თეორიული თვალსაზრისით უპირატესობა შეიძლება მიენიჭოს (a) ქვეპუნქტით გათვალისწინებულ ადეკვატურ პიკს, პრაქტიკაში უფრო ადვილია (b) ქვეპუნქტით გათვალისწინებული ადეკვატური პიკის განსაზღვრა.
პიკთა რაოდენობა განისაზღვრება იმ საანალიზო კომპონენტის ინფრაწითელ სპექტრში, რომლის სიხშირეებიც შეესაბამება დაკალიბრების სტანდარტის სპექტრის ადეკვატურ პიკს, ± 1სმ-1 ცდომილების ფარგლებში.
2.3.4.2. ინფრაწითელი სპექტრალური მონაცემების ინტერპრეტაცია
შთანთქმა წარმოდგენილი უნდა იქნეს საანალიზო კომპონენტის სპექტრის ყველა შრეში, რომელიც, თავის მხრივ, შეესაბამება დაკალიბრების სტანდარტის ეტალონური სპექტრის შესაბამის პიკ(ებ)ს. დაკალიბრების სტანდარტის ინფრაწითელ სპექტრში უნდა იყოს არანაკლებ ექვსი ადეკვატური პიკი. ექვსზე ნაკლები ადეკვატური პიკის შემთხვევაში, სპექტრის გამოყენება ეტალონურ სპექტრად დაუშვებელია. „ქულა“, ე.ი. ანალიზის დროს საანალიზო კომპონენტის ინფრაწითელ სპექტრში გამოვლენილი ადეკვატური პიკების პროცენტული მაჩვენებელი უნდა იყოს სულ მცირე 50. თუ არ არის ადექვატური პიკ(ებ)ის ზუსტი დამთხვევა, საანალიზო კომპონენტის სპექტრის სათანადო უბანი უნდა შეესაბამებოდეს ყველაზე შესაბამის პიკ(ებ)ს. აღნიშნული პროცედურა გამოიყენება მხოლოდ შთანთქმის პიკთან მიმართებაში ისეთ ნიმუშის სპექტრში, რომლის ინტენსივობა სამჯერ აღემატება ხმაურის სრულ ამპლიტუდას.
2.3.5. LC და სხვა მეთოდებით საანალიზო კომპონენტის განსაზღვრის სამუშაო კრიტერიუმები და სხვა მოთხოვნები
2.3.5.1. ქრომატოგრაფიული დაყოფა
შიდა სტანდარტი გამოყენებული უნდა იქნეს იმ შემთხვევაში, თუ აღნიშნული მიზნისათვის შესაბამისი მასალა ხელმისაწვდომია. სასურველია გამოყენებული იქნეს სტანდარტი, რომლის შეკავების დრო ახლოსაა საანალიზო კომპონენტის შეკავების დროსთან. საანალიზო კომპონენტის ელუცია (გამორეცხვა) ხდება ერთნაირ ექსპერიმენტულ პირობებში, შესაბამისი დაკალიბრების სტანდარტისათვის დამახასიათებელი შეკავების დროის განმავლობაში. საანალიზო კომპონენტის შეკავების მისაღები მინიმალური დრო ორჯერ უნდა აღემატებოდეს იმ შეკავების დროს, რომელიც შეესაბამება სვეტის თავისუფალ მოცულობას. საანალიზო კომპონენტის შეკავების დროის შეფარდება შიდა სტანდარტის შეკავების დროსთან, ანუ საანალიზო კომპონენტის შეკავების ფარდობითი დრო, უნდა იყოს იგივე, რაც დაკალიბრების სტანდარტის შეკავების დრო შესაბამის მატრიცაში, ± 2,5 %-ის ცდომილების ფარგლებში.
2.3.5.2. სრული სკანირება UV/VIS დეტექცია
დაცული უნდა იქნეს LC მეთოდის სამუშაო კრიტერიუმები.
საანალიზო კომპონენტის სპექტრში შთანთქმის მაქსიმალური მაჩვენებლები უნდა იყოს ტალღის იმავე სიგრძეზე, როგორიც დაკალიბრების სტანდარტის მაქსიმალური მაჩვენებლები, იმ ფარგლებში, რომლებიც განისაზღვრება დეტექციის სისტემის გარჩევადობით. დიოდური მატრიცის დეტექციისთვის, აღნიშნული, როგორც წესი, შეადგენს ± 2 ნმ-ს. საანალიზო კომპონენტი, რომლის სპექტრი 220 ნმ-ზე მეტია, ორი სპექტრის იმ ნაწილებისათვის, რომელთა შთანთქმის ფარდობითი უნარი ≥ 10%-ია, არ უნდა იყოს დაკალიბრების სტანდარტისაგან მკვეთრად განსხვავებული. ეს კრიტერიუმები დაკმაყოფილებულია თუ სახეზეა ერთნაირი მაქსიმალური მაჩვენებლები ან თუ ორ სპექტრს შორის განსხვავება, დაკვირვების ნებისმიერ წერტილში, არ აღემატება დაკალიბრების სტანდარტის შთანთქმის (აბსორბციის) 10%-ს.; თუ ძებნა და შერჩევა ხორციელდება კომპიუტერული ბიბლიოთეკის მონაცემთა ბაზის გამოყენებით, საკვლევ ნიმუშში სპექტრალური მონაცემების დაკალიბრების ხსნარის მონაცემებთან შედარება, უნდა აღემატებოდეს შესაბამისობის კრიტიკულ ფაქტორს. ამ ფაქტორის განსაზღვრა ხდება ვალიდაციის პროცესისას თითოეული საანალიზო კომპონენტისთვის იმ სპექტრების მიხედვით, რომლებისთვისაც სრულდება ზემოთ აღწერილი კრიტერიუმები. შემოწმებული უნდა იქნეს ნიმუშის მატრიცითა და დეტექტორის სამუშაო მახასიათებლებით გამოწვეული სპექტრ(ებ)ის ცვლილებები.
2.3.5.3. ფლუორომეტრიული გამოვლენის სამუშაო კრიტერიუმები
დაცული უნდა იქნეს LC მეთოდის სამუშაო კრიტერიუმები.
მათი გამოყენება უნდა მოხდეს იმ მოლეკულების მიმართ, რომელთაც ახასიათებთ ბუნებრივი ფლუორესცენცია ან მოლეკულების მიმართ, რომლებიც ფლუორესცენციის უნარს ამჟღავნებენ ტრანსფორმაციის ან დერივატიზაციის შემდეგ. აღგზნების ან ემისიის ეტაპებზე, ქრომატოგრაფიასთან ერთად, ტალღის სიგრძის შერჩევა უნდა მოხდეს იმგვარად, რომ მინიმუმამდე იქნეს შემცირებული ნიმუშის სუფთა ექსტრაქტში უცხო კომპონენტების წარმოქმნა.
ქრომატოგრამაში უახლოესი პიკის მაქსიმალური მაჩვენებელი, საანალიზო კომპონენტის განსაზღვრული პიკისგან გამოყოფილი უნდა იქნეს სულ მცირე საანალიზო კომპონენტის ერთი სრული პიკის სიგანით, საანალიზო კომპონენტის მაქსიმალური პიკის 10% სიმაღლის პირობებში.
2.3.5.4. LC-იმუნოგრამის მეშვეობით საანალიზო კომპონენტის განსაზღვრის სამუშაო კრიტერიუმები
LC იმუნოგრამის მეთოდი დამოუკიდებლად არ შეიძლება გამოყენებული იქნეს დამადასტურებელი მეთოდის სახით
დაცული უნდა იქნეს LC მეთოდის შესაბამისი კრიტერიუმები.
ხარისხის კონტროლის წინასწარ განსაზღვრული პარამეტრები, მაგ.: არასპეციფიკური ბმა, საკონტროლო ნიმუშების ფარდობითი კავშირი, სუფთა ნიმუშის შთანთქმის უნარის სიდიდე, უნდა იყოს ანალიზის ვალიდაციის დროს მიღებულ ფარგლებში;
იმუნოგრამა აგებული უნდა იქნეს სულ მცირე ხუთი ფრაქციის გამოყენებით.
თითოეული ფრაქცია უნდა იყოს პიკის სიგანის ნახევარზე ნაკლები.
ფრაქცია, რომელსაც გააჩნია საანალიზო კომპონენტის მაქსიმალური შემცველობა, უნდა იყოს ერთი და იგივე საეჭვო ნიმუშის, შეუსაბამო საკონტროლო ნიმუშისა და სტანდარტისათვის.
2.3.5.5. საანალიზო კომპონენტის განსაზღვრა LC მეთოდის გამოყენებით UV/VIS დეტექციასთან ერთად (ერთ ტალღის სიგრძეზე)
LC მეთოდი UV/VIS დეტექციასთან (ერთ ტალღის სიგრძეზე) ერთად დამოუკიდებლად არ შეიძლება გამოყენებული იქნეს დამადასტურებელი მეთოდის სახით
ქრომატოგრამაში უახლოესი პიკის მაქსიმალური მაჩვენებელი, საანალიზო კომპონენტის განსაზღვრული პიკისგან გამოყოფილი უნდა იქნეს სულ მცირე საანალიზო კომპონენტის ერთი სრული პიკის სიგანით, საანალიზო კომპონენტის მაქსიმალური პიკის 10% სიმაღლის პირობებში.
2.3.6. 2-DTLC მეთოდთან დაწყვილებული სრული სკანირებით UV/VIS სპექტრომეტრული დეტექციის მეთოდით საანალიზო კომპონენტის განსაზღვრის სამუშაო კრიტერიუმები და სხვა მოთხოვნები
სავალდებულოა ორგანზომილებიანი HPTLC და თანა-ქრომატოგრაფია.
საანალიზო კომპონენტის RF მნიშვნელობა სტანდარტის RF მნიშვნელობას უნდა შეესაბამებოდეს ±5 %-ის ფარგლებში.
საანალიზო კომპონენტი ვიზუალურად არ უნდა განსხვავდებოდეს სტანდარტისგან.
ერთი და იმავე ფერის ლაქების შემთხვევაში, უახლოესი ლაქის ცენტრი საანალიზო კომპონენტის ლაქის ცენტრისგან გამოყოფილი უნდა იქნეს სულ მცირე ლაქების დიამეტრების ჯამის ნახევარით.
საანალიზო კომპონენტის სპექტრი ვიზუალურად არ უნდა განსხვავდებოდეს სტანდარტის სპექტრისაგან, როგორც ეს დადგენილია სრული სკანირების UV/VIS დეტექციისთვის.
თუ ძებნა და შერჩევა ხორციელდება კომპიუტერული ბიბლიოთეკის მონაცემთა ბაზის გამოყენებით, საკვლევ ნიმუშში სპექტრალური მონაცემების დაკალიბრების ხსნარის მონაცემებთან თანაფარდობა უნდა აღემატებოდეს შესაბამისობის კრიტიკულ ფაქტორს. ამ ფაქტორის განსაზღვრა ხდება ვალიდაციის პროცესისას თითოეული საანალიზო კომპონენტისთვის იმ სპექტრების მიხედვით, რომლებისთვისაც სრულდება ზემოთ აღწერილი კრიტერიუმები. შემოწმებული უნდა იქნეს ნიმუშის მატრიცითა და დეტექტორის სამუშაო მახასიათებლებით გამოწვეული სპექტრ(ებ)ის ცვლილებები.
2.3.7. GC მეთოდისა და ელექტრონების დაჭერის დეტექციის (ECD) მეთოდით საანალიზო კომპონენტის განსაზღვრის სამუშაო კრიტერიუმები და სხვა მოთხოვნები
შიდა სტანდარტი გამოყენებული უნდა იქნეს იმ შემთხვევაში, თუ აღნიშნული მიზნისათვის შესაბამისი მასალა ხელმისაწვდომია. სასურველია გამოყენებული იქნეს მონათესავე ნივთიერება, რომლის შეკავების დრო ახლოსაა საანალიზო კომპონენტის შეკავების დროსთან. საანალიზო კომპონენტის ელუცია ხდება იმ შეკავების დროის განმავლობაში, რომელიც დამახასიათებელია შესაბამისი დაკალიბრების სტანდარტისათვის იგივე ექსპერიმენტულ პირობებში. საანალიზო კომპონენტის შეკავების მისაღები მინიმალური დრო ორჯერ უნდა აღემატებოდეს იმ შეკავების დროს, რომელიც შეესაბამება სვეტის თავისუფალ მოცულობას. საანალიზო კომპონენტის შეკავების დროის შეფარდება შიდა სტანდარტის შეკავების დროსთან, ანუ საანალიზო კომპონენტის შეკავების ფარდობითი დრო, უნდა იყოს იგივე, რაც დაკალიბრების სტანდარტის შეკავების დრო შესაბამის მატრიცაში, ± 0,5 %-ის ცდომილების ფარგლებში. ქრომატოგრამაში უახლოესი პიკის მაქსიმალური მაჩვენებელი, საანალიზო კომპონენტის განსაზღვრული პიკისგან გამოყოფილი უნდა იქნეს სულ მცირე საანალიზო კომპონენტის ერთი სრული პიკის სიგანით, საანალიზო კომპონენტის მაქსიმალური პიკის 10% სიმაღლის პირობებში. დამატებითი ინფორმაციის მისაღებად შესაძლებელია გამოყენებული იქნეს თანა-ქრომატოგრაფია.
2.4. დამადასტურებელი მეთოდები ელემენტებისთვის
ქიმიური ელემენტებისათვის დამადასტურებელი ანალიზი დაფუძნებული უნდა იყოს არაორაზროვან იდენტიფიკაციაზე, ასევე ამ ელემენტისათვის დამახასიათებელი ფიზიკურ-ქიმიური თვისებების მეშვეობით ელემენტის ზუსტ რაოდენობრივ განსაზღვრაზე (მაგ.: ატომური მასა, ქიმიური ელემენტისათვის დამახასიათებელი შთანთქმული და გაცემული გამოსხივების ტალღის სიგრძე) სიდიდის დონეზე.
ქიმიური ელემენტების იდენტიფიკაციისათვის გამოიყენება შემდეგი მეთოდები ან მეთოდების კომბინაცია:
ცხრილი №7
შესაფერისი დამადასტურებელი მეთოდები ქიმიური ელემენტებისთვის
|
მეთოდი |
გასაზომი პარამეტრი |
|
დიფერენციალურ-იმპულსური ანოდური გახსნის კულომეტრული მეთოდი ატომური აბსორბციის სპექტრომეტრია |
ელექტრონული სიგნალი
|
|
ალი |
აბსორბციის ტალღის სიგრძე |
|
ჰიდრიდის წარმოექმნა |
აბსორბციის ტალღის სიგრძე |
|
ცივი ორთქლი |
აბსორბციის ტალღის სიგრძე |
|
ელექტროთერმული გაფრქვევა (გრაფიტის ღუმელი) |
აბსორბციის ტალღის სიგრძე |
|
ატომურ-ემისიური სპექტრომეტრია ინდუქციურად შეწყვილებული პლაზმა |
ემისიის ტალღის სიგრძე |
|
მას-სპექტრომეტია ინდუქციურად შეწყვილებული პლაზმა |
მასისა და მუხტის თანაფარდობა |
2.4.1. საერთო სამუშაო კრიტერიუმები და სხვა მოთხოვნები დამადასტურებელი მეთოდებისადმი
ეტალონური ან გამდიდრებული მასალა, რომელიც შეიცავს საანალიზო კომპონენტის ცნობილ რაოდენობას, დასაშვები ზღვარის ან ზღვრული მნიშვნელობის (შეუსაბამო საკონტროლო ნიმუშის) ფარგლებში, ასევე შესაბამისი საკონტროლო მასალა და სუფთა რეაქტივები გამოყენებული უნდა იქნეს ანალიზის მსვლელობის პროცედურების განმავლობაში, საანალიზო ნიმუშების თითოეულ პარტიასთან ერთად. საანალიზო ხელსაწყოში ექსტრაქტების შეყვანა ხდება შემდეგი თანმიმდევრობით: სუფთა რეაქტივი, შესაბამისი საკონტროლო ნიმუში, ნიმუშ(ებ)ი, რომელთა დადასტურებაც აუცილებელია, ისევ შესაბამისი საკონტროლო ნიმუში და ბოლოს, შეუსაბამო საკონტროლო ნიმუში. ამ თანმიმდევრობის ნებისმიერი ცვლილება უნდა იქნეს დასაბუთებული.
საანალიზო კომპონენტის განსაზღვრისათვის გამოყენებული ანალიზის მეთოდები, როგორც წესი, ხსნარების მისაღებად საჭიროებს ორგანული მატრიცის დაშლას. ეს შესაძლებელია მიღწეული იქნეს მიკროტალღური მინერალიზაციით, რომელიც ამცირებს გამოსაკვლევი საანალიზო კომპონენტის დაკარგვის ან/და დაბინძურების რისკს. ამისათვის გამოიყენება ტეფლონის მაღალი ხარისხის დეზინფიცირებული ჭურჭელი. თუ ხდება სველი ან მშრალი დაშლის სხვა მეთოდების გამოყენება, ხელმისაწვდომი უნდა იყოს დოკუმენტურად დადასტურებული ინფორმაცია, რათა გამოირიცხოს პოტენციური დანაკარგის ან დაბინძურების რისკი. ორგანული მატრიცის დაშლის ალტერნატივად, გარკვეულ შემთხვევებში, შესაძლებელია გამოყენებული იქნეს გამოყოფის პროცედურები (მაგ. ექსტრაქცია), რათა საანალიზო კომპონენტი გამოყოფილი იქნეს მატრიცის კომპონენტიდან ან/და საანალიზო ხელსაწყოში შეყვანის მიზნით მოხდეს საანალიზო კომპონენტის კონცენტრირება.
გარე ან/და სტანდარტული ნივთიერების დამატებით დაკალიბრებისას თავიდან უნდა იქნეს აცილებული ანალიზისათვის დაწესებული სამუშაო დიაპაზონის გადაჭარბება. გარე დაკალიბრების შემთხვევაში, სავალდებულოა, რომ დაკალიბრების სტანდარტები მომზადებული იქნეს ხსნარში, რომელიც თავისი შემადგენლობით ახლოსაა ნიმუშის ხსნართან. გამოიყენება ასევე ფონის შესწორება, თუ აღნიშნული საჭიროა სპეციალური ანალიზური ვითარებებისათვის.
2.4.2. დამატებითი სამუშაო კრიტერიუმები და სხვა მოთხოვნები ანალიზის რაოდენობრივი მეთოდებისადმი
2.4.2.1. რაოდენობრივი მეთოდების სანდოობა
ელემენტებისათვის დაშვებული ეტალონური მასალის (CRM) განმეორებითი ანალიზის შემთხვევაში, ექსპერიმენტულად განსაზღვრული საშუალო რაოდენობის გადახრა, დაშვებულ რაოდენობასთან შედარებით, არ უნდა აღემატებოდეს ± 10%-ს. თუ არ არის ხელმისაწვდომი ასეთი დაშვებული ეტალონური მასალა (CRM), დასაშვებია გაზომვის სანდოობა შეფასებულ იქნეს უცნობ ნიმუშებზე ელემენტის ცნობილი რაოდენობების დამატებების აღდგენით; ყურადღება უნდა მიექცეს იმ ფაქტს, რომ საანალიზო კომპონენტისაგან განსხვავებით, დამატებული ელემენტი რეალურ მატრიცაში არ არის ქიმიურად დაკავშირებული და ამიტომ ასეთი მიდგომით მიღებული შედეგი ნაკლებად ვალიდურია, ვიდრე დაშვებული ეტალონური მასალის (CRM) გამოყენებით მიღებულ შედეგები. აღდგენის მონაცემები მისაღებია მხოლოდ იმ შემთხვევაში, თუ აღნიშნული მოქცეულია სამიზნე სიდიდის ± 10 % ფარგლებში.
2.4.2.2. რაოდენობრივი მეთოდების სიზუსტე
შიდალაბორატორიული აღწარმოებადობის პირობებში ნიმუშის განმეორებითი ანალიზისას, საშუალო სიდიდის ვარიაციის კოეფიციენტი (CV) არ უნდა აღემატებოდეს მაჩვენებლებს:
ცხრილი №8
რაოდენობრივი მეთოდებისათვის ვარიაციის კოეფიციენტი (CV) ელემენტების მასური წილის დიაპაზონში
|
მასური წილი |
CV (%) |
|
≥ 10 µg/kg to 100 µg/kg |
20 |
|
> 100 µg/kg to 1 000 µg/kg |
15 |
|
≥ 1 000 µg/kg |
10 |
3.4.2. განსაკუთრებული მოთხოვნები დიფერენციალურ-იმპულსური ანოდური გახსნის კულომეტრული მეთოდისთვის (DPASV)
DPASV განსაზღვრამდე მნიშვნელოვანია ნიმუშში ორგანული ნივთიერებების სრული დაშლა. ვოლტამოგრამაზე, ორგანული მასალების არსებობის გამო, არ ჩანს ფართო სიგნალი. არაორგანული მატრიცის შემადგენელმა კომპონენტებმა შესაძლოა გავლენა იქონიონ DPASV-ში პიკების სიმაღლეზე. ამიტომ, რაოდენობრივი განსაზღვრა უნდა განხორციელდეს სტანდარტის დამატების მეთოდის მეშვეობით. ნიმუშის ხსნარის ტიპური ვოლტამოგრამის ნიმუშები წარმოდგენილი უნდა იყოს მეთოდთან ერთად.
2.4.4. განსაკუთრებული მოთხოვნები ატომურ-აბსორციული სპექტრომეტრიის მიმართ (AAS)
ეს მეთოდი ძირითადად მონოელემენტურია და, შესაბამისად, საჭიროებს ექსპერიმენტული პარამეტრების ოპტიმიზაციას იმის მიხედვით, თუ რომელი ელემენტის რაოდენობის განსაზღვრა ხდება. შესაძლებლობის შესაბამისად, შედეგები შემოწმებული უნდა იქნეს როგორც თვისობრივად, ასევე რაოდენობრივად. აღნიშნული უნდა განხორციელდეს აბსორბციის ალტერნატიული ხაზების მეშვეობით (საუკეთესო ვარიანტია ორი სხვადასხვა ხაზის შერჩევა); დაკალიბრების სტანდარტი უნდა მომზადდეს იმ ხსნარის მატრიცაში, რომელიც შეძლებისდაგვარად ზუსტად ემთხვევა ნიმუშის გასაზომ ხსნარს (მაგ.: მჟავის კონცენტრაცია ან მოდიფიკატორის შემადგენლობა). მაჩვენებლების შეუსაბამობის მინიმუმამდე შემცირების მიზნით, ყოველი რეაქტივი შეძლებისდაგვარად უნდა იყოს უმაღლესი სისუფთავის. ნიმუშის აორთქლების ან/და გაფრქვევისათვის შერჩეული რეჟიმიდან გამომდინარე, შესაძლებელია სხვადასხვა ტიპის AAS-ის შერჩევა.
2.4.4.1. განსაკუთრებული მოთხოვნები ალის გამოყენებით ჩატარებულ ატომურ-აბსორციული სპექტრომეტრიისადმი (AAS)
ყოველ ელემენტთან დაკავშირებით უნდა განხორციელდეს ინსტრუმენტული პარამეტრების ოპტიმიზაცია. უნდა შემოწმდეს აირის შემადგენლობა და ნაკადის სიჩქარე. აუცილებელია გამოყენებული იქნეს ნაკადის სიჩქარის კორექტორი, რათა თავიდან იქნეს აცილებული ფონური აბსორბციით გამოწვეული ინტერფერენცია. უცნობი მატრიცის შემთხვევაში უნდა განხორციელდეს შემოწმება, საჭიროა თუ არა ფონური შესწორება.
2.4.4.2. განსაკუთრებული მოთხოვნები გრაფიტის ღუმელის გამოყენებით ჩატარებულ ატომურ-აბსორციული სპექტრომეტრიისადმი (AAS)
ლაბორატორიაში არსებული დაბინძურება ულტრა-კვალის დონეზე გრაფიტის ღუმელში მუშაობისას, ხშირად ახდენს ზეგავლენას სისწორეზე. ამიტომ გამოყენებული უნდა იქნეს მაღალი სისუფთავის რეაქტივები, დეიონიზირებული წყალი და ინერტული პლასტმასის ჭურჭელი ნიმუშებთან და სტანდარტებთან მუშაობისას. თითოეული ელემენტისათვის უნდა მოხდეს ხელსაწყოს პარამეტრების ოპტიმიზაცია. განსაკუთრებით შემოწმებული უნდა იქნეს წინასწარი დამუშავებისა და გაფრქვევის პირობები (ტემპერატურა, დრო). უნდა შემოწმდეს მატრიცის ცვლილება;
იზოთერმული გაფრქვევის პირობებში (მაგ. განივად გახურებული გრაფიტის მილი, ინტეგრირებული LVOV პლატფორმასთან (8), შეამცირებს საანალიზო კომპონენტის გაფრქვევასთან დაკავშირებულ მატრიცის ზემოქმედებას. მატრიცის ცვლილებისა და ZEEMAN-ის ფონური კორექციის ერთობლიობით (9), დასაშვებია საკალიბრო მრუდის საშუალებით რაოდენობის განსაზღვრა, სტანდარტული წყალხსნარების გაზომვის საფუძველზე.
2.4.5. განსაკუთრებული მოთხოვნები ატომურ-აბსორბციული სპექტრომეტრის მიმართ, ჰიდრიდების ნაერთების წარმოქმნისას
ორგანული ნივთიერებები, რომლებიც შეიცავენ დარიშხანს, ბისმუტს, გერმანიუმს, ტყვიას, სტიბიუმს, სელენს, კალას და ტელურს, შესაძლებელია იყოს ძალზე მდგრადი და, შესაბამისად, ელემენტის სრული შემადგენლობისათვის სწორი შედეგების მისაღებად, საჭიროებენ ჟანგვით დაშლას. ამიტომ რეკომენდებულია მიკროტალღური დაშლა ან მაღალი წნევით განაცრიანება ძლიერი დაჟანგვის პირობებში. განსაკუთრებული სიფრთხილით უნდა მივადევნოთ თვალი ელემენტების სრულ და აღწარმოებად გარდაქმნას მათ შესაბამის ჰიბრიდებად.
დარიშხანის ჰიდრიდის წარმოქმნა მარილმჟავას ხსნარში NaBH4-სთან ერთად დამოკიდებულია დარიშხანის დაჟანგვის ხარისხზე (As III: სწრაფი წარმოქმნა, As V: წარმოქმნის შედარებით ხანგრძლივი პერიოდი). ნაკადის შეფრქვევის მეთოდის გამოყენებით As V განსაზღვრისას მგრძნობელობის დაკარგვის თავიდან ასაცილებლად, რომელიც გამოწვეულია ამ სისტემაში რეაქციის მოკლე დროით, As V, ჟანგვითი დაშლის შემდეგ უნდა გარდაიქმნას As III-ად. აღნიშნული მიზნებისათვის ხელსაყრელია კალიუმის იოდიდი/ასკორბინის მჟავა ან ცისტეინი. სუფთა ნიმუშების, დაკალიბრების ხსნარებისა და ნიმუშების ხსნარების დამუშავება უნდა მოხდეს ერთი და იმავე მეთოდით. პარტიის სისტემასთან მუშაობა შესაძლებლობას იძლევა დარიშხანის ორივე ნაერთი განისაზღვროს სისწორეზე რაიმე გავლენის მოხდენის გარეშე. As V ჰიდრიდის შენელებული წარმოქმნის გამო, აუცილებელია დაკალიბრება შესრულდეს პიკების ზონების ინტეგრირებით. უნდა მოხდეს ხელსაწყოს პარამეტრების ოპტიმიზაცია. განსაკუთრებით მნიშვნელოვანია და საჭიროებს შემოწმებას აირის ნაკადი, რომელსაც ჰიდრიდი გადააქვს ატომიზატორისკენ.
2.4.6. განსაკუთრებული მოთხოვნები ცივი ორთქლით ჩატარებული ატომურ-აბსორბციული სპექტრომეტრის მიმართ
ცივი ორთქლი გამოიყენება მხოლოდ ვერცხლისწყლის შემთხვევაში. ელემენტარული ვერცხლისწყლის აქროლადობითა და ადსორბციით გამოწვეული დანაკარგის გამო, ანალიზის მსვლელობისას აუცილებელია განსაკუთრებული ყურადღება. თავიდან უნდა იქნეს აცილებული გარემოთი და რეაქტივებით გამოწვეული დაბინძურება.
ორგანული ნაერთები, რომლებიც ვერცხლისწყალს შეიცავენ, ვერცხლისწყლის საერთო შემცველობის განსაზღვრისათვის, საჭიროებენ ჟანგვით დაშლას. დაშლისათვის გამოყენებული უნდა იქნეს მიკროტალღური დაშლის ან მაღალი წნევით განაცრიანების დახურული სისტემები. განსაკუთრებული სიფრთხილეა საჭირო იმ მოწყობილობების გარეცხვისას, რომლებიც შეხებაშია ვერცხლისწყალთან.
ხელსაყრელია ნაკადის შეფრქვევის მეთოდით მუშაობა. უფრო დაბალი ზღვრული მნიშვნელობისთვის რეკომენდებულია ელემენტარული ვერცხლისწყლის ადსორბცია ოქროს/პლატინის ადსორბენტზე, რასაც მოსდევს თერმული დესორბცია. თავიდან უნდა იქნეს აცილებული ადსორბენტის ან უჯრედის კონტაქტი სინესტესთან, რამდენადაც ეს მოქმედებს გაზომვის მაჩვენებელზე.
2.4.7. განსაკუთრებული მოთხოვნები ინდუქციურად შეწყვილებული პლაზმით ჩატარებული ატომურ-ემისიური სპექტრომეტრიის (ICP-AES) მიმართ
ინდუქციურად შეწყვილებული პლაზმით ჩატარებული ატომურ-ემისიური სპექტრომეტრია (10) წარმოადგენს მრავალ-ელემენტურ მეთოდს, რომელიც სხვადასხვა ელემენტების ერთდროულ გაზომვის საშუალებას იძლევა. ICP-AES გამოყენებისათვის თავდაპირველად უნდა მოხდეს ნიმუშების დაშლა, რათა განხორციელდეს ორგანული მატრიცების შემადგენელ ნაწილებად დაშლა. გამოყენებული უნდა იქნეს მიკროტალღური დაშლის ან მაღალი წნევით განაცრიანების დახურული სისტემები. ICP-AES ანალიზისათვის მნიშვნელოვან როლს ასრულებს ინსტრუმენტის და ელემენტის დაკალიბრება ან ტალღის სიგრძის შერჩევა. ხელსაწყოს დაკალიბრებისათვის, დაკალიბრების ხაზოვანი მრუდის შემთხვევაში, აუცილებელია მხოლოდ ოთხი კონცენტრაციის დაკალიბრების ხსნარის გაზომვა, რამდენადაც ICP-AES-ის საკალიბრო მრუდები, როგორც წესი, სწორხაზოვანია კონცენტრაციის ოთხი-ექვსი მნიშვნელობისთვის. ICP-AES სისტემის დაკალიბრება ხდება მრავალელემენტიანი სტანდარტის გამოყენებით, რომელიც მზადდება ხსნარში, რომელსაც იგივე მჟავური კონცენტრაცია აქვს, როგორიც გასაზომ ხსნარს. ხაზოვანი მრუდისათვის მოწმდება ელემენტების კონცენტრაციები.
ტალღის სიგრძის შერჩევა საანალიზო კომპონენტის ემისიის გაზომვისათვის მიზანშეწონილია მოხდეს განსასაზღვრი ელემენტების კონცენტრაციებისთვის. იმ შემთხვევაში, როდესაც საანალიზო კომპონენტის კონცენტრაცია სცილდება ემისიის ხაზის სამუშაო დიაპაზონს, გამოიყენება ემისიის სხვა ხაზი. თავდაპირველად შერჩეული უნდა იქნეს ყველაზე მგრძნობიარე ემისიის ხაზი (დაბრკოლებების გარეშე), შემდეგ ნაკლებად მგრძნობიარე. გამოვლენის ზღვარზე ან მასთან ახლოს მყოფ ზღვართან მუშაობისას, საანალიზო კომპონენტისათვის საუკეთესოა ყველაზე მგრძნობიარე ხაზი. სპექტრალური და ფონური დაბრკოლებები იწვევენ ICP-AES გამოყენებისას სირთულეებს. დაბრკოლებები შესაძლებელია იყოს უბრალო ფონური გადაადგილება, ფონის დახრის გადანაცვლება, პირდაპირი სპექტრალური გადაფარვა და კომპლექსური ფონური გადაადგილება. თითოეულ ამ დაბრკოლებას აქვს თავისი მიზეზები და თავიდან აცილების გზები. მატრიცის მიხედვით ხდება დაბრკოლებების კორექცია და სამუშაო პარამეტრების ოპტიმიზაცია. ზოგიერთი დაბრკოლება შესაძლებელია თავიდან იქნეს აცილებული მატრიცების გაზავებით ან ადაპტაციის გზით. საკვლევი ნიმუშების თითოეული პარტიის შემთხვევაში აუცილებელია საკვლევი ნიმუშის მსგავსად დამუშავდეს ის ეტალონური და გამდიდრებული მასალა, რომელიც შეიცავს საანალიზო კომპონენტის ცნობილ რაოდენობას, აგრეთვე სუფთა მასალაც. გადახრის გამოკვლევისათვის უნდა მოხდეს სტანდარტის შემოწმება, მაგ.: 10 სინჯის შემდეგ. ყველა რეაქტივი და პლაზმური აირი უნდა იყოს ყველაზე მაღალი სისუფთავის.
2.4.8. განსაკუთრებული მოთხოვნები ინდუქციურად შეწყვილებული პლაზმით ჩატარებული ატომურ-ემისიური სპექტრომეტრიის (ICP-AES)(11)) მიმართ
საშუალო ატომური მასის მქონე მიკროელემენტების როგორიცაა ქრომი, სპილენძი და ნიკელი, გამოვლენაზე შესაძლოა დიდი გავლენა იქონიოს სხვა იზობარულმა ან პოლიატომურმა იონებმა. ამის აცილება შესაძლებელია მხოლოდ იმ შემთხვევაში, თუ გარჩევადობის უნარის სიმძლავრე არანაკლებ 7 000-8 000-ია. MS მეთოდთან დაკავშირებული სირთულეები მოიცავს ხელსაწყოების მაჩვენებლების გადახრას, მატრიცის ზემოქმედებას და მოლეკულური იონების ჩარევას (m/z < 80). ხელსაწყოების მონაცემების გადახრისა და მატრიცის ზემოქმედების კორექციისათვის სავალდებულოა მრავლობითი შიდა სტანდარტიზაცია, რომელიც მოიცავს განსასაზღვრი ელემენტების მასის დიაპაზონების იდენტურ დიაპაზონს.
ICP-MS გაზომვამდე საჭიროა ორგანული ნივთიერებების სრული დაშლა. AAS-ს მეთოდის ანალოგიურად, ჰერმეტულ ჭურჭელში დაშლის შემდეგ აქროლადი ელემენტები, მაგ. იოდი, სტაბილიზირებული უნდა იყოს დაჟანგვით. ყველაზე ძლიერია არგონის (პლაზმური აირი), წყალბადის, ნახშირბადის, აზოტის და ჟანგბადის მოლეკულური იონების (ძლიერი მჟავები, პლაზმური აირებისა და ატმოსფერული აირების მინარევები) და ნიმუში მატრიცის კომბინაციის შედეგად წარმოქმნილი ზემოქმედება. დაბრკოლებების თავიდან აცილებისათვის აუცილებელია სრული დაშლა, ფონური გაზომვები, შესაბამისი საანალიზო მასის შერჩევა, რომელიც ზოგჯერ დაკავშირებულია ნაკლებ რაოდენობასთან (დეტექციის დაბალი ზღვარი) და ძლიერი მჟავების სათანადო შერჩევა, მაგ. აზოტმჟავა.
განსასაზღვრ ელემენტებთან დაკავშირებით გამორიცხული უნდა იქნეს დაბრკოლებები, სპეციალური ანალიზური მასების შესაბამისი შერჩევით, მათ შორის იზოტოპების თანაფარდობების დადასტურებით. თითოეული გაზომვისათვის შიდა სტანდარტების გამოყენებით უნდა მოხდეს ხელსაწყოების მახასიათებლების შემოწმება Fano-ფაქტორების გათვალისწინებით.
3. ვალიდაცია
ვალიდაცია აჩვენებს, რომ ანალიზის მეთოდი შეესაბამება შესაბამისი სამუშაო მახასიათებლების კრიტერიუმებს.
კონტროლის განსხვავებული მიზნები სხვადასხვა მეთოდებს საჭიროებს. ქვემოთ მოცემული ცხრილი ადგენს თუ რომელი სამუშაო მახასიათებელი უნდა დადასტურდეს და რომელი მეთოდისთვის.
ცხრილი №9
ანალიზის მეთოდების კლასიფიკაცია იმ სამუშაო მახასიათებლების მიხედვით, რომლებიც უნდა განისაზღვროს
|
დეტექციის ლიმიტი CCß |
ზღვრული მნიშვნელობა CCα |
სანდოობა/აღდგენა |
სიზუსტე |
შერჩევითობა/ სპეციფიკურობა |
გამოყენებადობა/ მდგრადობა/ სტაბილურობა |
||
|
თვისობრივი მეთოდები |
S |
+ |
– |
– |
– |
+ |
+ |
|
C |
+ |
+ |
– |
– |
+ |
+ |
|
|
რაოდენობრივი მეთოდები |
S |
+ |
– |
– |
+ |
+ |
+ |
|
C |
+ |
+ |
+ |
+ |
+ |
+ |
|
S = სკრინინგის მეთოდები; C = დამადასტურებელი მეთოდები; + = სავალდებულოა განსაზღვრა.
3.1. ვალიდაციის პროცედურები
ამ თავში მოცემულია ანალიზის მეთოდების ვალიდაციის პროცედურების მაგალითები ან/და მითითებები. შესაძლებელია სხვა მეთოდების გამოყენებაც იმის საჩვენებლად, რომ კონკრეტული ანალიზის მეთოდი შეესაბამება სამუშაო მახასიათებლებისთვის სამუშაო კრიტერიუმებს, იმ პირობით, რომ მიღწეული იქნება ინფორმაციის იგივე დონე და ხარისხი.
ვალიდაცია აგრეთვე შესაძლებელია განხორციელდეს კოდექს ალიმენტარიუსის (Codex Alimentarius), ISO-ს ან IUPAC-ის(12) მიერ დადგენილი ლაბორატორიათაშორისი გამოკვლევის ჩატარების გზით, ან ალტერნატიული მეთოდით, როგორიცაა ერთ ლაბორატორიაში ჩატარებული კვლევა ან შიდა ვალიდაცია(13)(14). ამ ნაწილში ყურადღება გამახვილებულია ერთ ლაბორატორიაში ჩატარებულ კვლევებზე (შიდა ვალიდაციაზე) მოდულარული მიდგომის გამოყენებით. ეს მიდგომა მოიცავს:
1. გამოყენებული ვალიდაციის მოდელისგან დამოუკიდებლად მდგარ საერთო სამუშაო მახასიათებლების ნაკრებს და
2. მოდელზე დამოკიდებულ, უფრო სპეციფიურ პროცედურებს, როგორც ეს აღწერილია ცხრილ №10-ში.
ცხრილი №10
მოდელისგან დამოუკიდებელი და მოდელზე დამოკიდებული სამუშაო მახასიათებლები
|
ვალიდაცია |
||
|
მოდელისგან დამოუკიდებელი სამუშაო პარამეტრები |
მოდელზე დამოკიდებული სამუშაო პარამეტრები |
|
|
საერთო სამუშაო მახასიათებლები (3.1.1) |
ტრადიციული ვალიდაციის მეთოდი (3.1.2) |
შიდა ვალიდაციის მეთოდი (3.1.3) |
|
სპეციფიკურობა |
აღდგენა |
აღდგენა |
|
სანდოობა |
განმეორებადობა |
განმეორებადობა |
|
მდგრადობა: უმნიშვნელო ცვლილებებით |
შიდალაბორატორიული აღწარმოებადობა |
შიდალაბორატორიული აღწარმოებადობა |
|
სტაბილურობა |
აღწარმოებადობა |
აღწარმოებადობა |
|
ზღვრული მნიშვნელობა (CCα) |
ზღვრული მნიშვნელობა (CCα) |
|
|
გამოვლენის შესაძლებლობა (CCβ) |
გამოვლენის შესაძლებლობა (CCβ) |
|
|
დაკალიბრების მრუდები |
დაკალიბრების მრუდები |
|
|
მდგრადობა: მნიშვნელოვანი ცვლილებები |
მდგრადობა |
|
3.1.1. მოდელისგან დამოუკიდებელი სამუშაო მახასიათებლები
ვალიდაციისათვის შერჩეული მეთოდის მიუხედავად, საჭიროა დადგენილი იქნეს შემდეგი სამუშაო მახასიათებლები. სამუშაოს მოცულობის შესამცირებლად, შესაძლებელია გამოყენებული იქნეს გულდასმით შემუშავებული და სტატისტიკურად სანდო მიდგომა, რათა შესაძლებელი გახდეს ჩატარებული ექსპერიმენტების გაერთიანება სხვადასხვა პარამეტრების დასადგენად.
3.1.1.1. სპეციფიკურობა
ანალიზის მეთოდებისთვის მნიშვნელოვანია საანალიზო კომპონენტსა და მასთან მჭიდროდ დაკავშირებულ ნივთიერებებს (იზომერები, მეტაბოლიტები, დაშლის პროდუქტები, ენდოგენური ნივთიერებები, მატრიცის შემადგენლები და ა.შ) შორის განსხვავებების დადგენის უნარი. დაბრკოლებების შემოწმებისათვის აუცილებელია ორი მეთოდი.
შესაბამისად, უნდა მოხდეს პოტენციურად დამაბრკოლებელი ნივთიერებების ამორჩევა და შესაბამისი სუფთა ნიმუშების ანალიზი, რათა გამოვლინდეს შესაძლო დაბრკოლებების არსებობა და შეფასდეს მათი ზემოქმედება:
– შეარჩიეთ ქიმიურად დაკავშირებული ნაერთები (მეტაბოლიტები, დერივატივები, და სხვ.) ან სხვა ნივთიერებები, რომლებიც გვხვდება გამოსაკვლევ ნაერთთან ერთად და რომლებიც შეიძლება იყოს წარმოდგენილი ნიმუშში;
– გაანალიზეთ შესაბამისი რეპრეზენტატული სუფთა ნიმუშების რაოდენობა (n ≥ 20) და შეამოწმეთ შესაძლო დაბრკოლებების არსებობა (სიგნალები, პიკები, იონების კვალი) კვლევის იმ მიდამოში, სადაც მოსალოდნელია სამიზნე საანალიზო კომპონენტის ელუცია;
– დამატებით, შესაბამისი რეპრეზენტატული სუფთა ნიმუშები გამდიდრებული უნდა იქნეს იმ ნივთიერების შესაბამისი კონცენტრაციით, რომელიც სავარაუდოდ ხელს შეუშლის საანალიზო კომპონენტის იდენტიფიცირებას ან/და რაოდენობის განსაზღვრას;
– ანალიზის შემდგომ გამოიკვლიეთ:
– შეიძლება თუ არა დაბრკოლების არსებობამ გამოიწვიოს მცდარი იდენტიფიცირება;
– აბრკოლებს თუ არა ერთი ან მეტი დაბრკოლების არსებობა გამოსაკვლევი საანალიზო კომპონენტის იდენტიფიცირებას; ან
– ახდენს თუ არა მნიშვნელოვან ზეგავლენას რაოდენობის განსაზღვრაზე.
3.1.1.2. სანდოობა
ამ აბზაცში აღწერილია სანდოობის (სისწორის ერთი კომპონენტის) განსაზღვრა. სანდოობა შეიძლება დადგინდეს მხოლოდ დაშვებული ეტალონური მასალის მეშვეობით (CRM). CRM გამოყენებული უნდა იქნეს შეძლებისდაგვარად. პროცედურა დეტალურად განისაზღვრება ISO 5725-4-ში (5); ქვემოთ მოცემულია მაგალითი:
– უნდა განხორციელდეს CRM-ს ექვსი ასლის ანალიზი აღნიშნული მეთოდის ტესტირების ინსტრუქციების შესაბამისად;
– უნდა განისაზღვროს საანალიზო კომპონენტის კონცენტრაცია ყოველი ასლის ნიმუშში;
– გამოთვლილი უნდა იქნეს აღნიშნული კონცენტრაციების საშუალო, სტანდარტული გადახრა და ვარიაციის კოეფიციენტი (%);
– სანდოობა გამოითვლება გამოვლენილი საშუალო კონცენტრაციის გაყოფით დაშვებულ მნიშვნელობაზე (რომელიც იზომება როგორც კონცენტრაცია) და 100-ზე გამრავლებით, რათა შედეგი გამოხატული იქნეს პროცენტული მაჩვენებლით;
– სანდოობა (%) = აღდგენით კორექტირებული გამოვლენილი საშუალო კონცენტრაცია × 100/დაშვებული მნიშვნელობა.
CRM-ის არარსებობის შემთხვევაში, სანდოობის ნაცვლად აღდგენა შესაძლოა განისაზღვროს 4.1.2.1 პუნქტით დადგენილი წესით.
3.1.1.3. გამოყენებადობა/მდგრადობა (უმნიშვნელო ცვლილებები)
მსგავსი კვლევების დროს ხდება ლაბორატორიის მიერ უმნიშვნელო დასაბუთებული ცვლილებების შეტანა და მათ შედეგებზე დაკვირვება.
გამოკვლევის დაწყებამდე უნდა განხორციელდეს წინასწარი გამოკვლევა, რომლის დროსაც ხდება ნიმუშის იმ წინასწარი დამუშავების, გასუფთავებისა და ანალიზის ფაქტორების შერჩევა, რომლებმაც შესაძლოა ზეგავლენა იქონიოს გაზომვის შედეგებზე. ამგვარი ფაქტორები შესაძლოა მოიცავდეს საანალიზო კომპონენტს, ასევე რეაქტივების წყაროსა და ასაკს, გამხსნელებს, სტანდარტებს და ნიმუშების ექსტრაქტებს, გაცხელების სიჩქარეს, ტემპერატურას, pH მნიშვნელობას, და მრავალ სხვა ფაქტორს, რომლებსაც შესაძლოა ადგილი ჰქონდეს ლაბორატორიაში. აღნიშნული ფაქტორების ცვლილება უნდა განხორციელდეს იმ სიდიდით, რომელიც ემთხვევა ლაბორატორიაში ჩვეულებრივ გამოვლენილ გადახრებს. მათ შორის:
– უნდა განხორციელდეს იმ შესაძლო ფაქტორთა იდენტიფიცირება, რომლებმაც შესაძლოა ზეგავლენა იქონიონ შედეგებზე;
– თითოეული ფაქტორის ცვლილება უნდა განხორციელდეს მცირედით;
– მდგრადობის ტესტი უნდა ჩატარდეს Youden-ის მეთოდის გამოყენებით(15)(16). (აღნიშნულ შემთხვევაში შესაძლებელია სხვა აღიარებული მეთოდების გამოყენებაც, თუმცა, Youden-ის მეთოდი საჭიროებს მინიმალურ დროსა და ძალისხმევას). Youden-ის მეთოდი წარმოადგენს ფაქტორულ ექსპერიმენტს. სხვადასხვა ფაქტორებს შორის ურთიერთქმედება ვერ დგინდება;
– იმ შემთხვევაში, თუ აღმოჩნდა, რომ ფაქტორი მნიშვნელოვან ზეგავლენას ახდენს გაზომვის შედეგებზე, აღნიშნული ფაქტორის მისაღებობის ფარგლების განსაზღვრის მიზნით უნდა ჩატარდეს დამატებითი ექსპერიმენტები;
– ფაქტორები, რომლებიც მნიშვნელოვან ზეგავლენას ახდენენ შედეგებზე, მკაფიოდ უნდა იქნენ იდენტიფიცირებულნი მეთოდის ოქმში.
მიზანი მდგომარეობს არა თითო ჯერზე თითო ცვლილების შესწავლაში, არამედ ერთდროულად რამდენიმე ვარიაციის შეტანაში. მაგალითად: დავუშვათ, შვიდი სხვადასხვა ფაქტორი, რომლებმაც შესაძლოა ზეგავლენა იქონიონ შედეგებზე გამოხატული იყოს A, B, C, D, E, F, G ნომინალური მნიშვნელობებით, იმ შემთხვევაში თუ მათი ნომინალური ცვლილებები უმნიშვნელოდ შეიცვლება. დავუშვათ, მათი ალტერნატიული მნიშვნელობები გამოხატული იყოს შესაბამისი პატარა ასოებით a, b, c, d, e, f და g. აღნიშნულის შედეგად ვიღებთ 27 ანუ 128 სხვადასხვა შესაძლო კომბინაციას.
შესაძლებელია შერჩეულ იქნეს რვა კომბინაცია, რომელთაც აქვთ ბალანსი ლათინურ დიდ და პატარა ასოებს შორის (ცხრილი №11) უნდა განხორციელდეს რვა განსაზღვრა, რომლებშიც გამოყენებული იქნება შერჩეულ ფაქტორების (A-G) კომბინაციები. ამ განსაზღვრის შედეგები მოცემულია ცხრილ №11-ში, როგორც S-Z.
ცხრილი №11
მდგრადობის (უმნიშვნელო ცვლილებების) შესწავლის ექსპერიმენტის გეგმა
|
ფაქტორული მნიშვნელობა F |
განსაზღვრის კომბინაციების რაოდენობა |
|||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
|
A/a
B/b
C/c
D/d
E/e
F/f
G/g |
A
B
C
D
E
F
G |
A
B
c
D
e
f
g |
A
b
C
d
E
f
g |
A
b
c
d
e
F
G |
a
B
C
d
e
F
g |
a
B
c
d
E
f
G |
a
b
C
D
e
f
G |
A
b
c
D
E
F
g |
|
მიღებული შედეგი R |
S |
T |
U |
V |
W |
X |
Y |
Z |
კალკულაციებისათვის იხ. მდგრადობაზე ტესტების მაგალითები 3.3-ში.
3.1.1.4. სტაბილურობა
შენახვისას ან ანალიზის დროს ნიმუშში საანალიზო კომპონენტის ან მატრიცის კომპონენტების არასაკმარისმა სტაბილურობამ შესაძლოა ანალიზის შედეგებში გამოიწვიოს მნიშვნელოვანი გადახრა. ამასთანავე, შემოწმებული უნდა იქნეს ხსნარში დაკალიბრების სტანდარტის სტაბილურობა. ჩვეულებრივ საანალიზო კომპონენტის სტაბილურობა კარგად ხასიათდება შენახვის სხვადასხვა პირობებში. შენახვის პირობების მონიტორინგი წარმოადგენს ლაბორატორიის აკრედიტაციის სისტემის ნაწილს. როდესაც სტაბილურობა არ არის ცნობილი იგი შეიძლება განისაზღვროს ქვემოთ მოცემული მაგალითების შესაბამისად.
საანალიზო კომპონენტის სტაბილურობის განსაზღვრა ხსნარში ხდება შემდეგნაირად:
– უნდა მომზადდეს საანალიზო კომპონენტის ახალი ხსნარი და ტესტირების ინსტრუქციების შესაბამისად მოხდეს მისი განზავება თითოეული შერჩეული კონცენტრაციის შესაბამისი ჯერადობით (aliquot) (მაგ.: 40). (იმ ნივთიერებებისათვის, რომელთა დასაშვები ზღვარი არ არის დადგენილი – დაახლოებით მინიმალური აუცილებელი სამუშაო ზღვარი, ან სხვა ნივთიერებებისათვის – დაახლოებით დაშვებული ზღვარი). უნდა მომზადდეს საანალიზო კომპონენტის ორივე ხსნარი, რომელიც გამოიყენება გამდიდრებისათვის და ხსნარის საბოლოო ანალიზისათვის ან ნებისმიერი სხვა გამოსაკვლევი ხსნარი (მაგ.: წარმოებული სტანდარტები);
– ახლად მომზადებულ ხსნარში საანალიზო კომპონენტის შემადგენლობა უნდა შემოწმდეს ტესტირების ინსტრუქციების შესაბამისად;
– შესაბამისი მოცულობები უნდა გადანაწილდეს შესაბამის კონტეინერებში, გაუკეთდეს ეტიკეტები და შენახულ იქნეს შემდეგი სქემის მიხედვით:
ცხრილი №12
ხსნარში საანალიზო კომპონენტის სტაბილურობის განსაზღვრის სქემა
|
– 20 °C |
+ 4 °C |
+ 20 °C |
|
|
მუქი
ღია |
10 ჯერადობა |
10 ჯერადობა |
10 ჯერადობა
10 ჯერადობა |
– შენახვის ვადად შესაძლოა შეირჩეს ერთი, ორი, სამი ან ოთხი კვირა, ან უფრო ხანგრძლივი ვადა საჭიროებისამებრ, მაგ.: იდენტიფიკაციის ან/და რაოდენობის განსაზღვრის დროს პირველი დეგრადაციის ნიშნების გამოჩენამდე. აღრიცხული უნდა იქნეს შენახვის მაქსიმალური ვადა და შენახვის ოპტიმალური პირობები;
– ყოველ ჯერადში საანალიზო კომპონენტების კონცენტრაციის გამოთვლა უნდა განხორციელდეს ანალიზის დროს ახლად მომზადებული საანალიზო კომპონენტის ხსნარის 100% გამოყენებით;
მითითება გრაფიკულ გამოსახულებაზე
Ci = კონცენტრაცია დროის მოცემული მომენტისათვის
Cfresh = ახალი ხსნარის კონცენტრაცია
მატრიცაში საანალიზო კომპონენტის სტაბილურობა
– როდესაც შესაძლებელია, კვლევისას გამოყენებული უნდა იქნეს გამოსაკვლევი ნიმუშები (incurred samples). როდესაც ასეთი ნიმუშები არ არის ხელმისაწვდომი, გამოყენებული უნდა იქნეს საანალიზო კომპონენტით გამდიდრებული მატრიცა;
– გამოსაკვლევი მასალის არსებობის შემთხვევაში, კონცენტრაცია აღნიშნულ მასალაში უნდა განისაზღვროს მანამ, სანამ მასალა ჯერ კიდევ ახალია. მასალის შემდგომი ჯერადების აღება შესაძლებელია ერთი, ორი, ოთხი და 20 კვირის შემდეგ და უნდა განისაზღვროს კონცენტრაციები. ქსოვილის შენახვა უნდა განხორციელდეს სულ მცირე -20 ºC ან, საჭიროების შემთხვევაში, უფრო დაბალ ტემპერატურაზე;
– თუ გამოსაკვლევი მასალა არ არის ხელმისაწვდომი, აღებულ უნდა იქნეს სუფთა მასალა, უნდა მოხდეს მისი ჰომოგენიზაცია. მასალა გაყოფილი უნდა იქნეს ხუთ ჯერად ნაწილად. თითოეული ჯერადი ნაწილი გამდიდრებული უნდა იქნეს საანალიზო კომპონენტით, რომელიც სასურველია მომზადდეს მცირე რაოდენობით წყალხსნარში. ერთი ჯერადი ნაწილის ანალიზი უნდა განხორციელდეს დაუყონებლივ. დარჩენილი ჯერადი ნაწილები შენახული უნდა იქნეს სულ მცირე -20 ºC ან, თუ საჭიროა, უფრო დაბალ ტემპერატურაზე და ჩატარდეს მისი ანალიზი ერთი, ორი, ოთხი ან ოცი კვირის შემდეგ.
3.1.1.5. საკალიბრო მრუდები
რაოდენობის განსაზღვრისას საკალიბრო მრუდების გამოყენების შემთხვევაში:
– მრუდის ასაგებად გამოყენებული უნდა იქნეს სულ მცირე ხუთი დონე (ნულის ჩათვლით);
– აღწერილი უნდა იქნეს საკალიბრო მრუდის სამუშაო დიაპაზონი;
– აღწერილი უნდა იქნეს მრუდის მათემატიკური ფორმულა და მონაცემების შესაბამისობა მრუდთან,
– აღწერილი უნდა იქნეს საკალიბრო მრუდის პარამეტრების მისაღებობის დიაპაზონი;
როდესაც აუცილებელია სტანდარტულ ხსნარზე დაფუძნებული სერიული კალიბრაცია, მითითებული უნდა იქნეს საკალიბრო მრუდის პარამეტრების მისაღებობის დიაპაზონი, რომელიც შესაძლოა განსხვავდებოდეს სერიების მიხედვით.
3.1.2. ვალიდაციის ტრადიციული პროცედურები
ტრადიციული მეთოდებით პარამეტრების გამოთვლა საჭიროებს რამდენიმე ინდივიდუალური ექსპერიმენტის ჩატარებას. ყოველ მნიშვნელოვან ცვლილებასთან დაკავშირებით განსაზღვრული უნდა იქნეს სამუშაო მახასიათებლები (იხ. ზემოთ „გამოყენებადობა/მდგრადობა“ ნაწილში). მულტი-ანალიტური მეთოდებისათვის უნდა განხორციელდეს რამდენიმე საანალიზო კომპონენტის ერთდროული ანალიზი; შესაძლებლობის მიხედვით უნდა მოხდეს შესაბამისი დაბრკოლებების წინასწარ გამორიცხვა. რამდენიმე სამუშაო მახასიათებელი შესაძლებელია განისაზღვროს იდენტური გზით. სამუშაოს მოცულობის მინიმუმამდე შესამცირებლად რეკომენდებულია შეძლებისდაგვარად, რაც შეიძლება მეტი ექსპერიმენტის გაერთიანება (მაგ.: განმეორებადობისა და შიდალაბორატორიული აღწარმოებადობის გაერთიანება სპეციფიკურობასთან, სუფთა ნიმუშების ანალიზთან, ზღვრული მნიშვნელობისა და სპეციფიკურობის განსაზღვრისათვის).
3.1.2.1. აღდგენა
CRM-ის არარსებობის შემთხვევაში, აღდგენა უნდა განისაზღვროს ექსპერიმენტების მეშვეობით, რომელთა დროსაც გამოყენებული იქნება გამდიდრებული სუფთა მატრიცა შემდეგი სქემის მიხედვით:
– შეირჩეს სუფთა მასალის 18 ჯერადი ნაწილი და გამდიდრდეს ექვსის ჯერადი თითოეული 1, 1.5 და 2 ჯერ, მინიმალურ აუცილებელ სამუშაო ზღვარზე ან დასაშვებ ზღვარზე 0,5, 1 და 1.5- ჯერ;
– განხორციელდეს ნიმუშების ანალიზი და ყოველ ნიმუშში განისაზღვროს კონცენტრაცია;
– თითოეული ნიმუშისათვის განისაზღვროს აღდგენა ქვემოთ მოცემული განტოლების შესაბამისად;
– უნდა გამოითვალოს აღდგენის საშუალო მაჩვენებელი და CV თითოეული დონის ექვსი შედეგიდან;
– აღდგენა % = 100 x გაზომილი შემცველობა/გამდიდრების დონე.
ეს ტრადიციული მეთოდი აღდგენის განსაზღვრებასთან დაკავშირებით წარმოადგენს ამ წესის 34-ე მუხლით განსაზღვრულ სტანდარტის დამატების მეთოდის ვარიანტს, როდესაც:
– ნიმუში საანალიზო ნიმუშის ნაცვლად განიხილება სუფთა ნიმუშად;
– ითვლება, რომ შედეგი (1) და აღდგენა (2) ორ საკვლევ ნაწილში მსგავსია;
– საკვლევ ნიმუშებს აქვთ ერთნაირი მასა და საკვლევი ნაწილიდან ხდება ერთნაირი მოცულობების ექსტრაქცია;
– ყოველ მეორე (დანამატებიან) საკვლევ ნაწილზე დამატებული დაკალიბრების სტანდარტის რაოდენობა აღინიშნება როგორც xADD. (xADD=ρA.VA);
– x1 წარმოადგენს სუფთა ნიმუშის გაზომილ მნიშვნელობას, ხოლო x2 – მეორე (დანამატებიანი) საკვლევი ნაწილის გაზომილ მნიშვნელობას;
– მაშინ, აღდგენა % = 100 (x2 – x1)/xADD.
როდესაც ერთ-ერთი ზემოაღნიშნული პირობა არ უნდა იქნეს მიღწეული (ან მიჩნეულია, რომ არ უნდა იქნეს მიღწეული), მაშინ აღდგენის საშუალო მაჩვენებლის განსაზღვრის მთლიანი პროცედურა უნდა განხორციელდეს 3.5-ში აღწერილი სტანდარტის დამატების მეთოდით.
3.1.2.2. განმეორებადობა
– უნდა მომზადდეს საანალიზო კომპონენტით გამდიდრებული იდენტური მატრიცების ნიმუშების ნაკრები, რათა მიღებულ იქნეს კონცენტრაციები, რომლებიც ეკვივალენტურია მინიმალური აუცილებელი სამუშაო ზღვარის 1, 1.5 და 2-ზე ნამრავლზე ან დასაშვები ზღვარის 0,5, 1 და 1.5-ზე ნამრავლზე.
– თითოეულ დონეზე ანალიზი უნდა ჩატარდეს სულ მცირე ექვსჯერ.
– უნდა განხორციელდეს ნიმუშების ანალიზი.
– გამოთვლილ იქნეს თითოეულ ნიმუშში გამოვლენილი კონცენტრაცია.
– გამოთვლილი უნდა იქნეს გამდიდრებული ნიმუშების საშუალო კონცენტრაცია, სტანდარტული გადახრა და ვარიაციის კოეფიციენტი (%);
– აღნიშნული უნდა განმეორდეს სულ მცირე ორ სხვა შემთხვევაში.
– უნდა გამოითვალოს საერთო საშუალო კონცენტრაცია და CV გამდიდრებულ ნიმუშებთან მიმართებაში.
3.1.2.3. შიდალაბორატორიული აღწარმოებადობა
– უნდა მომზადდეს საანალიზო კომპონენტით გამდიდრებული განსაზღვრული საკვლევი მასალის (იდენტური ან განსხვავებული მატრიცების) ნიმუშების ნაკრები, რათა მიღებულ იქნეს კონცენტრაციები, რომლებიც ეკვივალენტურია მინიმალური აუცილებელი სამუშაო ზღვარის 1, 1.5 და 2-ზე ნამრავლზე ან დასაშვები ზღვარის 0,5, 1 და 1.5-ზე ნამრავლზე.
– თითოეულ დონეზე ანალიზი უნდა ჩატარდეს სულ მცირე ექვსჯერ.
– ეს მოქმედება უნდა განმეორდეს სულ მცირე ორ სხვა შემთხვევაში, თუ შესაძლებელია, სხვადასხვა ოპერატორთან და განსხვავებულ გარემო-პირობებში, მაგ.: რეაქტივების, გამხსნელების და ა.შ. სხვადასხვა პარტია, ოთახის სხვადასხვა ტემპერატურა, სხვადასხვა ინსტრუმენტები და ა.შ.
– უნდა განხორციელდეს ნიმუშების ანალიზი.
– გამოთვლილ იქნეს თითოეულ ნიმუშში გამოვლენილი კონცენტრაცია.
– გამოთვლილი უნდა იქნეს გამდიდრებული ნიმუშების საშუალო კონცენტრაცია, სტანდარტული გადახრა და ვარიაციის კოეფიციენტი (%).
3.1.2.4. აღწარმოებადობა
აღწარმოებადობის დასადასტურებლად ლაბორატორიებმა მონაწილეობა უნდა მიიღონ ერთობლივ კვლევებში ISO 5725-2-ის (5) შესაბამისად.
3.1.2.5. ზღვრული მნიშვნელობა (CCα))
ზღვრული მნიშვნელობა უნდა დადგინდეს იმ მოთხოვნების შესაბამისად, რომლებიც განსაზღვრულია იდენტიფიკაციისთვის ან იდენტიფიკაციას პლიუს რაოდენობის განსაზღვრისთვის, როგორც ეს მითითებულია პუნქტში – „სამუშაო კრიტერიუმები და სხვა მოთხოვნები ანალიზის მეთოდებისადმი“ (მე-2 ნაწილი).
იმ ნივთიერებებთან მიმართებაში, რომლებისთვისაც არ არის დადგენილი დასაშვები ზღვარი, CCα-ს დადგენა ხდება ერთ-ერთი შემდეგი პროცედურით:
– ISO 11843-ის(17) მიხედვით ჩატარებული საკალიბრო მრუდის პროცედურის შესაბამისად (აქ მითითებულია, როგორც სუფთა მდგომარეობის ცვლადის კრიტიკული მნიშვნელობა). აღნიშნულ შემთხვევაში გამოიყენება სუფთა მასალა, რომელიც გამდიდრებული უნდა იქნეს მინიმალურ აუცილებელ სამუშაო დონემდე და ზევით თანაბარი ინტერვალებით. უნდა განხორციელდეს ნიმუშების ანალიზი. იდენტიფიკაციის შემდგომ უნდა მოხდეს გრაფიკის სახით სიგნალის დამოკიდებულების აღნიშვნა დამატებულ კონცენტრაციასთან მიმართებაში. შესაბამისი კონცენტრაცია კოორდინატთა y ღერძის გადაკვეთის წერტილში, პლიუს გადაკვეთის წერტილის შიდალაბორატორიული აღწარმოებადობის სტანდარტული გადახრა გამრავლებული 2,33-ზე, ტოლია ზღვრული მნიშვნელობის. ეს გამოიყენება მხოლოდ რაოდენობრივი ანალიზის ჩატარებისას (α = 1%);
– ან თითოეულ მატრიცაზე სულ მცირე 20 სუფთა მასალის ანალიზით, რათა შესაძლებელი იყოს სიგნალ-ხმაურის თანაფარდობის დროის იმ მონაკვეთში, რომელშიც მოსალოდნელია საანალიზო კომპონენტი. ზღვრულ მნიშვნელობად შეიძლება გამოყენებულ იქნეს სიგნალ-ხმაურის თანაფარდობა გამრავლებული სამზე. აღნიშნული გამოიყენება რაოდენობრივი და თვისობრივი ანალიზის დროს.
იმ ნივთიერებებთან მიმართებაში, რომლებისთვისაც დადგენილია დასაშვები ზღვარი, CCα-ს დადგენა ხდება ერთ-ერთი შემდეგი პროცედურით:
– ISO 11843-ს(17) მიხედვით ჩატარებული საკალიბრო მრუდის პროცედურის შესაბამისად (აქ მითითებულია, როგორც სუფთა მდგომარეობის ცვლადის კრიტიკული მნიშვნელობა). აღნიშნულ შემთხვევაში გამოიყენება სუფთა მასალა, რომელიც გამდიდრებული უნდა იქნეს მინიმალურ აუცილებელ სამუშაო დონემდე და ზევით თანაბარი ინტერვალებით. უნდა განხორციელდეს ნიმუშების ანალიზი. იდენტიფიკაციის შემდგომ უნდა მოხდეს გრაფიკის სახით სიგნალის დამოკიდებულების აღნიშვნა დამატებულ კონცენტრაციასთან მიმართებაში. შესაბამისი კონცენტრაცია დაშვებულ ზღვარზე, პლიუს შიდალაბორატორიული აღწარმოებადობის სტანდარტული გადახრა გამრავლებული 1,64-ზე, ტოლია ზღვრული მნიშვნელობის (α = 5%);
– თითოეულ მატრიცაზე სულ მცირე 20 სუფთა მასალის ანალიზით, რომლებიც გამდიდრებულია საანალიზო კომპონენტით დასაშვებ ზღვრამდე. შესაბამისი კონცენტრაცია დაშვებულ ზღვარზე, პლიუს სტანდარტული გადახრა გამრავლებული 1,64-ზე, ტოლია ზღვრული მნიშვნელობის (α = 5%).
იხ. ასევე მე-5 მუხლი და 3.2 პუნქტი.
3.1.2.6. გამოვლენის შესაძლებლობა (CCβ)
გამოვლენის შესაძლებლობა უნდა განისაზღვროს იმ მოთხოვნების შესაბამისად, რომლებიც დადგენილია სკრინინგისთვის, იდენტიფიკაციისთვის ან იდენტიფიკაციას პლიუს რაოდენობრივი მაჩვენებელის განსაზღვრისთვის (იხ. მე-2 ნაწილი).
იმ ნივთიერებებთან მიმართებაში, რომლებისთვისაც არ არის დადგენილი დასაშვები ზღვარი, CCβ-ს დადგენა ხდება ერთ-ერთი შემდეგი პროცედურით:
– ISO 11843-ს(17) მიხედვით ჩატარებული საკალიბრო მრუდის პროცედურის შესაბამისად (აქ მითითებულია, როგორც სუფთა მდგომარეობის ცვლადის მინიმალური გამოვლენადი მნიშვნელობა). აღნიშნულ შემთხვევაში გამოიყენება სუფთა მასალა, რომელიც გამდიდრებული უნდა იქნეს მინიმალურ აუცილებელ სამუშაო დონემდე და ქვევით თანაბარი ინტერვალებით. უნდა განხორციელდეს ნიმუშების ანალიზი. იდენტიფიკაციის შემდგომ უნდა მოხდეს გრაფიკის სახით სიგნალის დამოკიდებულების აღნიშვნა დამატებულ კონცენტრაციასთან მიმართებაში. შესაბამისი კონცენტრაცია ზღვრულ მნიშვნელობაზე, პლიუს 1,64-ჯერ საშუალო გაზომილი შემცველობის შიდალაბორატორიული აღწარმოებადობის სტანდარტული გადახრა ზღვრულ მნიშვნელობაზე, ტოლია გამოვლენის შესაძლებლობის (β = 5%);
– თითოეულ მატრიცაზე სულ მცირე 20 სუფთა მასალის ანალიზით, რომლებიც გამდიდრებულია საანალიზო კომპონენტით ზღვრულ მნიშვნელობამდე. უნდა მოხდეს ნიმუშების ანალიზი და საანალიზო კომპონენტის იდენტიფიცირება. ზღვრული მნიშვნელობის სიდიდეს დამატებული 1,64- ჯერ გაზომილი შემცველობის შიდალაბორატორიული აღწარმოებადობის სტანდარტული გადახრა ტოლია გამოვლენის შესაძლებლობის (β = 5%);
– თუ არ არსებობს რაოდენობრივი შედეგები, გამოვლენის შესაძლებლობა შესაძლებელია განისაზღვროს გამდიდრებული სუფთა მასალის გამოკვლევით, ზემოაღნიშნულ ზღვრულ მნიშვნელობაზე ან მის ზემოთ. აღნიშნულ შემთხვევაში, კონცენტრაციის დონე, სადაც რჩება მხოლოდ ≤ 5% ცრუ დადებითი შედეგი, ტოლია მეთოდის გამოვლენის შესაძლებლობისა. შესაბამისად, უნდა ჩატარდეს სულ მცირე 20 კვლევა კონცენტრაციის სულ მცირე ერთ დონესთან მიმართებაში, რათა უზრუნველყოფილი იქნეს აღნიშნული განსაზღვრის საიმედო საფუძველი.
იმ ნივთიერებებთან მიმართებაში, რომლებისთვისაც დადგენილია დასაშვები ზღვარი, CCâ-ს დადგენა ხდება ერთ-ერთი შემდეგი პროცედურით:
– ISO 11843-ს(17) მიხედვით ჩატარებული საკალიბრო მრუდის პროცედურის შესაბამისად (აქ მითითებულია, როგორც სუფთა მდგომარეობის ცვლადის მინიმალური გამოვლენადი მნიშვნელობა). აღნიშნულ შემთხვევაში გამოიყენება რეპრეზენტატული სუფთა მასალა, რომელიც გამდიდრებული უნდა იქნეს დაახლოებით დასაშვებ დონემდე თანაბარი ინტერვალებით. უნდა მოხდეს ნიმუშების ანალიზი და საანალიზო კომპონენტის იდენტიფიცირება. გამოანგარიშებული უნდა იქნეს ზღვრულ მნიშვნელობაზე გაზომილი საშუალო შემცველობის სტანდარტული გადახრა. შესაბამისი კონცენტრაცია დაშვებული ზღვარის სიდიდეზე, პლიუს შიდალაბორატორიული აღწარმოებადობის სტანდარტული გადახრა გამრავლებული 1,64-ზე, ტოლია გამოვლენის შესაძლებლობისა (â = 5%),
– თითოეულ მატრიცაზე სულ მცირე 20 სუფთა მასალის ანალიზით, რომლებიც გამდიდრებულია საანალიზო კომპონენტით ზღვრულ მნიშვნელობამდე. ზღვრული მნიშვნელობის სიდიდე, პლიუს შესაბამისი სტანდარტული გადახრა გამრავლებული 1,64-ზე, ტოლია გამოვლენის შესაძლებლობისა (â = 5%).
იხ. ასევე ნაწილი 3.2.
3.1.2.7. მდგრადობა (მნიშვნელოვანი ცვლილებები)
ანალიზის მეთოდის ტესტირება უნდა განხორციელდეს სხვადასხვა ექსპერიმენტული პირობებში, რომელიც მოიცავს მაგ.: სხვადასხვა ნაერთებს, სხვადასხვა მატრიცებს ან ნიმუშის აღების სხვადასხვა პირობებს. შეტანილი ცვლილებები უნდა იყოს მნიშვნელოვანი. აღნიშნული ცვლილებების მნიშვნელობა შესაძლებელია შეფასდეს, მაგალითად, Youden-ის მეთოდის გამოყენებით (15) (16). თითოეული სამუშო მახასიათებელი უნდა განისაზღვროს ყველა იმ მნიშვნელოვან ცვლილებებთან მიმართებაში, რომლებმაც მნიშვნელოვანი გავლენა მოახდინა ანალიზის შესრულებაზე.
3.1.3. ვალიდაცია ალტერნატიული მოდელების შესაბამისად
ალტერნატიული ვალიდაციის პროცედურების გამოყენებისას, ვალიდაციის ოქმში შეტანილი უნდა იქნეს საბაზისო მოდელი და სტრატეგია შესაბამის წინაპირობებთან, ვარაუდებთან და ფორმულებთან ერთად, ან როგორც მინიმუმ, მითითებული უნდა იქნეს მათი ხელმისაწვდომობა. ქვემოთ მოცემულია ალტერნატიული მიდგომის მაგალითი. შიდა ვალიდაციის მოდელის გამოყენებისას, სამუშაო მახასიათებლები განისაზღვრება იმგვარად, რომ შესაძლებელია მნიშვნელოვანი ცვლილებების დადასტურება იგივე ვალიდაციის პროცედურის ფარგლებში. ეს მოითხოვს ვალიდაციისთვის ექსპერიმენტული გეგმის შემუშავებას.
3.1.3.1. ექსპერიმენტული გეგმა
ექსპერიმენტული გეგმა უნდა შემუშავდეს გამოსაკვლევი სხვადასხვა ნაერთებისა და სხვადასხვა ფაქტორების რაოდენობის მიხედვით. შესაბამისად, ვალიდაციის მთლიანი პროცედურის პირველ ეტაპზე განისაზღვრება საკვლევი პოპულაციები, რომელთა ანალიზიც უნდა განხორციელდეს შემდგომში ლაბორატორიაში, იმისათვის, რათა შეირჩეს ყველაზე მნიშვნელოვანი სახეობები და ის ფაქტორები, რომლებმაც შესაძლებელია გავლენა იქონიონ გაზომვის შედეგებზე. შესაბამისად, კონცენტრაციის დიაპაზონი შერჩეული უნდა იქნეს მიზნიდან გამომდინარე, მნიშვნელობის დონის (level of interest) შესაბამისად.
მაგალითი:
– შესაძლებელია რამდენიმე საანალიზო კომპონენტის გამოკვლევა იმ ანალიზის მეთოდის პარალელურად, რომლის ვალიდაციაც ხდება;
– განისაზღვრა წამყვანი ფაქტორის ორი ვარიანტი (A და B). მთავარი ფაქტორები წარმოქმნიან საფუძველს, რომლის მიხედვითაც ერთიანდება ფაქტორის დონეები. აღნიშნული მთავარი ფაქტორები შესაძლოა მოიცავდნენ ისეთ ფაქტორებს, როგორიცაა სახეობები ან მატრიცა. აღნიშნულ მაგალითში მთავარი ფაქტორი მერყეობდა ორ დონეზე, ანუ ორი სხვადასხვა სახეობა (სახეობა A და B) იქნა განხილული. ზოგადად, შესაძლებელია მთავარი ფაქტორების ვარირება ორზე მეტ დონეზე, რომელიც მხოლოდ ზრდის ჩასატარებელი ანალიზების რაოდენობას;
– შერჩეული ფაქტორები უნდა ვარირებდნენ ორ დონეზე (მითითებული როგორც “ + „ ან „–“).
ცხრილი №13
ვალიდაციის პროცედურისათვის მნიშვნელოვნად მიჩნეული ფაქტორების მაგალითები
|
ცხოველი სქესი ჯიში ტრანსპორტირების პირობები შენახვის პირობები ნიმუშის სიახლე მოსუქების პირობები სხვადასხვა ოპერატორი სხვადასხვა გამოცდილებით |
(ფაქტორი 1) (ფაქტორი 2) (ფაქტორი 3) (ფაქტორი 4) (ფაქტორი 5) (ფაქტორი 6) (ფაქტორი 7) |
ცხრილი №14
შესაძლო ექსპერიმენტული გეგმა ზემოთ მოყვანილი მაგალითისთვის
|
სახეობა |
ფაქტორი 1 |
ფაქტორი 2 |
ფაქტორი 3 |
ფაქტორი 4 |
ფაქტორი 5 |
ფაქტორი 6 |
ფაქტორი 7 |
ნიმუშის ნომერი |
|
A
A
A
A
A
A
A
A
A
A |
+
+
+
+
–
–
–
– |
+
+
–
–
+
+
–
– |
+
–
+
–
+
–
+
– |
+
–
–
+
–
+
+
– |
–
+
–
+
+
–
+
– |
+
–
–
+
+
–
–
+ |
–
–
+
+
+
+
–
– |
1
2
3
4
5
6
7
8 |
|
B
B
B
B
B
B B
B
B
B |
+
+
+
+
–
–
–
– |
+
+
–
–
+
+
–
– |
+
–
+
–
+
–
+
– |
+
–
–
+
–
+
+
– |
+
–
+
–
–
+
–
+ |
–
+
+
–
–
+
+
– |
+
+
–
–
–
–
+
+ |
9
10
11
12
13
14
15
16 |
რამდენადაც თითოეული ნიმუში (ფაქტორის დონის თითოეული კომბინაცია) დაკავშირებული უნდა იყოს ოთხ სხვადასხვა კონცენტრაციასთან მნიშვნელობის დონესთან ახლოს, და თითოეული დონისთვის ანალიზი უნდა ჩაუტარდეს ერთ სუფთა ნიმუშს, სრული ვალიდაციის ექსპერიმენტის განხორციელებისათვის უნდა ჩატარდეს 5 x 16 = 80 ანალიზი.
აღნიშნული 80 გაზომვის შედეგიდან შესაძლებელია გამოითვალოს (13) (14)
აღდგენა
– კონცენტრაციის თითოეული დონის განმეორებადობა (Sir);
– კონცენტრაციის თითოეული დონის შიდალაბორატორიული აღწარმოებადობა (Sir);
– ზღვრული მნიშვნელობა (CCα);
– გამოვლენის შესაძლებლობა (CCβ);
– სიმძლავრის მრუდი (β-ცდომილება კონცენტრაციის წინააღმდეგ (იხ. 3.1.3.2);
– მნიშვნელოვანი ცვლილებებისადმი მდგრადობა; უმნიშვნელო ცვლილებებისადმი მდგრადობა შეიძლება განისაზღვროს 3.1.1.3 აბზაცის მიხედვით;
– 16 ნიმუშთან დაკავშირებული საკალიბრო მრუდი;
– ერთი საერთო საკალიბრო მრუდი;
– საერთო საკალიბრო მრუდის საპროგნოზო ინტერვალი;
– მატრიცით გამოწვეული გადახრები (Smat);
– ანალიზის პროცესით გამოწვეული გადახრები (Srun);
– ინდივიდუალური ფაქტორების ზეგავლენა გაზომვის შედეგებზე.
აღნიშნული სამუშაო მახასიათებლები მეთოდის ეფექტურობის კომპლექსური შეფასების შესაძლებლობას იძლევა, ვინაიდან გამოკვლეულია არა მხოლოდ ცალკეული ფაქტორების ზეგავლენა, არამედ მათი შესაბამისი კომბინაციები. ექსპერიმენტის გეგმის დახმარებით შესაძლებელია მიღებულ იქნეს გადაწყვეტილება საერთო საკალიბრო მრუდიდან ამა თუ იმ შერჩეული ფაქტორის გამორიცხვის შესახებ, ვინაიდან აღნიშნული მნიშვნელოვნად განსხვავდება სხვა ფაქტორების სტანდარტული გადახრიდან.
3.1.3.2. სიმძლავრის მრუდი
სიმძლავრის მრუდი იძლევა ინფორმაციას შერჩეული კონცენტრაციის დიაპაზონის ფარგლებში იმ გამოვლენის შესაძლებლობის შესახებ, რომელიც ამ მეთოდს გააჩნია. აღნიშნული ეხება გამოსაკვლევი მეთოდის გამოყენებისას β – ცდომილების რისკს. სიმძლავრის მრუდი საშუალებას იძლევა გამოთვლილ იქნეს გამოვლენის შესაძლებლობა მეთოდების შესაბამისი კატეგორიებისთვის (სკრინინგი, დადასტურება), ან სახეობებისთვის (ხარისხობრივი თუ რაოდენობრივი) გარკვეულ β –ცდომილებასთან მიმართებაში (მაგ.: 5%);
გრაფიკი 1
სიმძლავრის მრუდი

გრაფიკი 1 გვიჩვენებს ანალიზის მეთოდის გამოვლენის შესაძლებლობის (Ccβ) გრაფიკულ შესრულებას. ამ კონკრეტულ მეთოდში რჩება მცდარი გადაწყვეტილების მიღების რისკი, რომელიც ტოლია 5%-ისა 0,50 µგ/კგ კონცენტრაციაზე. 0.55 მკგ/კგ კონცენტრაციაზე კი ცრუ დადებითი გადაწყვეტილების მიღების ცდომილება მცირდება 1%-მდე.
3.1.3.3. აღწარმოებადობა
მეთოდის აღწარმოებადობის განსაზღვრა ერთ ლაბორატორიაში ჩატარებული კვლევებით (შიდა ვალიდაცია) საჭიროებს ISO 43-1(3) და 43-2(4) ინსტრუქციით გათვალისწინებულ პროფესიულ სწავლებებში რეგულარულ მონაწილეობას. ლაბორატორიებს ეძლევათ საკუთარი მეთოდების შერჩევის შესაძლებლობა იმ პირობით, რომ აღნიშნული მეთოდები გამოყენებული იქნება ჩვეულ პირობებში. ლაბორატორიის სტანდარტული გადახრა შესაძლოა გამოყენებული იქნეს მეთოდის აღწარმოებადობის შეფასებისათვის.
3.2. სხვადასხვა ანალიზური კვლევების შედეგად მიღებული ზღვრების გრაფიკული გამოსახულებები
გრაფიკი 2
ნივთიერებები, რომელთათვისაც არ არის დადგენილი დასაშვები ზღვარი
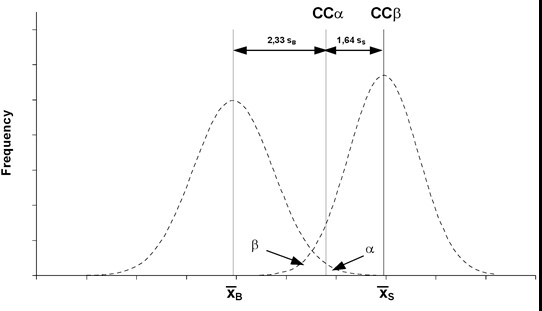
რეაგირება
XS – დაბინძურებული ნიმუშის საშუალო რეაგირების მნიშვნელობა;
SB – სუფთა ნიმუშის სტანდარტული გადახრა (განსაზღვრულია ლაბორატორიის შიდა აღწარმოებადობის პირობებში);
SS – დაბინძურებული ნიმუშის სტანდარტული გადახრა (განსაზღვრულია ლაბორატორიის შიდა აღწარმოებადობის პირობებში);
α – არასწორი შეუსაბამო შედეგების ნორმა;
β – არასწორი შესაბამისი შედეგების ნორმა;
CCα – რეაგირება მოცემული α შეცდომით და 50% β შეცდომით;
CC β – რეაგირება ძალიან მცირე α შეცდომით და β შეცდომით.
გრაფიკი 3
ნივთიერებები, რომელთათვისაც დადგენილია დასაშვები ზღვარი

XB – სუფთა ნიმუშის საშუალო „კონცენტრაცია“;
XPL – დასაშვებ დონეზე ანალიტის შემცველი ნიმუშის საშუალო კონცენტრაცია;
XS – დაბინძურებული ნიმუშის საშუალო კონცენტრაცია;
SPL – დასაშვებ დონეზე ანალიტის შემცველი ნიმუშის სტანდარტული გადახრა (განსაზღვრულია ლაბორატორიის შიდა აღწარმოებადობის პირობებში);
SS – შემცველი ნიმუშის სტანდარტული გადახრა (განსაზღვრულია ლაბორატორიის შიდა აღწარმოებადობის პირობებში);
α – არასწორი შეუსაბამო შედეგების ნორმა;
β – არასწორი შესაბამისი შედეგების ნორმა;
CCα – რეაგირება მოცემული α შეცდომით და 50% β შეცდომით;
CCβ – რეაგირება ძალიან მცირე α შეცდომით და β შეცდომით.
3.3. უმნიშვნელო ცვლილებების შეტანით ჩატარებული მდგრადობის კვლევის შედეგების გამოთვლის მაგალითი Youden-ის (16) მეთოდის მიხედვით
საშუალო მნიშვნელობების შედარება (A)
|
AA = AB = |
Σ(Ai)/4 Σ(Bi)/4 |
შეადარეთ დიდი ასოების საშუალო მნიშვნელობები (AA to AG) მათი შესაბამის პატარა ასოების საშუალო მნიშვნელობებთან (Aa to Ag). თუ ფაქტორს გააჩნია გავლენა, მაშინ განსხვავება იქნება ბევრად დიდი, ვიდრე სხვა ფაქტორების განსხვავებები. |
|
AC = |
Σ(Ci)/4 |
|
|
AD =
AE = |
Σ(Di)/4
Σ(Ei)/4 |
მდგრად მეთოდზე გავლენა არ უნდა მოახდინოს ლაბორატორიებს შორის დიდი ალბათობით არსებულმა ცვლილებებმა. |
|
AF = |
Σ(Ei)/4 |
თუ სახეზეა არ არის მნიშვნელოვანი განსხვავება, ყველაზე რეალური საზომი შემთხვევითი ცდომილებისა მოცემულია შვიდი განსხვავებით. |
|
AG = |
Σ(Gi)/4 |
|
|
Aa = |
Σ(ai)/4 |
|
|
Ab = |
Σ(bi)/4 |
|
|
Ac = |
Σ(ci)/4 |
|
|
Ad = |
Σ(di)/4 |
|
|
Ae = |
Σ(ei)/4 |
|
|
Af = |
Σ(fi)/4 |
|
|
Ag = |
Σ(gi)/4 |
|
განსხვავებები (Di) |
განსხვავებების კვადრატი (Di) |
|
Da = A – a = Σ(Ai) – Σ(ai)
Db = B – b = Σ(Bi) – Σ(bi)
Dc = C – c = Σ(Ci) – Σ(ci)
Dd = D – d = Σ(Di) – Σ(di)
De = E – e = Σ(Ei) – Σ(ei)
Df = F – f = Σ(Fi) – Σ(fi)
Dg = G – g = Σ(Gi) – Σ(gi) |
Da2 = value a
Db2 = value b
Dc2 = value c
Dd2 = value d
De2 = value e
Df2 = value f
Dg2 = value g |
განსხვავებების სტანდარტული გადახრა Di (SDi):
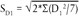
როდესაც SDi მნიშვნელოვნად აღემატება იმ მეთოდის სტანდარტულ გადახრას, რომელიც ტარდება ლაბორატორიათაშორისი აღწარმოებადობის პირობებში (იხ. ზემოთ) დანამდვილებით შეგვიძლია ვთქვათ, რომ ყველა ფაქტორი ერთობლივად ახდენს შედეგზე გავლენას, მაშინაც კი თუ ცალკეული ფაქტორი არ ახდენს მნიშვნელოვან ზეგავლენას და რომ ეს მეთოდი არ არის საკმარისად სანდო შერჩეული მოდიფიკაციებისთვის.
3.4. შიდა ვალიდაციის პროცედურის გამოთვლის მაგალითები
შიდა ვალიდაციის პროტოკოლის მაგალითები და გამოთვლები, როგორც ეს აღწერილია ალტერნატიული მოდელების მიხედვით ჩატარებულ ვალიდაციაში (3.1.3) (13) (14).
3.5. სტანდარტის დამატების მეთოდის მაგალითები
საკვლევი ნიმუში, რომელიც შეიცავს T საანალიზო კომპონენტს, იყოფა ორ – 1 და 2 – საკვლევ ნაწილად, შესაბამისად m1 და m2 მასით. საკვლევი ნაწილი 2 უკავშირდება საანალიზო კომპონენტის ρA კონცენტრაციის ხსნარის VA მოცულობას. საკვლევი ნიმუშის ორი ექსტრაქტი, V1 და V2 შესაბამისი მოცულობით, მიიღება ექსტრაქციითა და გასუფთავებით. სავარაუდოდ, საანალიზო კომპონენტის აღდგენა უნდა იყოს rc. ორივე ექსტრაქტის ანალიზი ხორციელდება b მგრძნობელობის გაზომვის მეთოდით და აღნიშნული იძლევა შესაბამისად X1 და X2 ანალიზურ სიგნალს.
თუ დაშვებულია, რომ საანალიზო კომპონენტის თავდაპირველი და მინარევებიანი ნიმუშისათვის rc და b იდენტურია, შესაძლებელი იქნება T-ს შემცველობა გამოანგარიშებული იქნეს შემდეგნაირად:
T = x1.V1.ρA.VA/(x2.V2.m1- x1.V1.m2)
მეთოდი იძლევა rc აღდგენის განსაზღვრის შესაძლებლობას ზემოაღწერილ ანალიზზე დამატებით 1 საკვლევი ნიმუშის ექსტრაქტის ნაწილს (მოცულობა V3) ემატება საანალიზო კომპონენტის ρB .VB ცნობილი რაოდენობა და ხდება მისი ანალიზი. ანალიზის პასუხი ტოლია X3 -ის და აღდგენა უდრის:
rc = x2.V1.V2.ρB.VB/[x3.V1.V3(T.m2+ρA.VA) - x2.V2.T.m1(V3-VB)]
გარდა ამისა, შესაძლებელია b მგრძნობელობის გამოანგარიშებაც როგორც:
b=x1.V1/rc.T.m1
აღწერილია გამოყენების ყველა პირობა და დეტალი (18).
4. გამოყენებული აბრევიატურები
AAS (atomic absorption spectrometry) – ატომურ-აბსორბციული სპექტომეტრია
AES (atomic emission spectrometry) – ატომურ-ემისიური სპექტომეტრია
AOAC-I (Association of Official Analytical Chemists INTERNATIONAL) – სახელმწიფო ორგანიზაციებში მომუშავე ანალიტიკოს-ქიმიკოსთა საერთაშორისო ასოციაცია
B bound fraction (immunoassays) – შეკავშირებული ფრაქცია (იმუნოლოგიური ანალიზი)
CI (chemical ionisation) – ქიმიური იონიზაცია
CRM (certified reference material) – ატესტირებული (დაშვებული) ეტალონური მასალა
CV (coefficient of variation) – ვარიაციის კოეფიციენტი
2D (two dimensional) – ორგანზომილებიანი
DAD (diode array detection) – დიოდური გამოვლენა
DPASV (differential pulse anodic stripping voltametry) – დიფერენციალურ-იმპულსური ანოდური გახსნის კულომეტრული მეთოდი
ECD (electron capture detection) – გამოვლენა ელექტრონების დაჭერით
EI (electronic impact ionisation) – ელექტრონის დარტყმით იონიზაცია
GC (gas chromatography) -აირ-ქრომატოგრაფია
HPLC (high performance liquid chromatography) – მაღალეფექტური სითხური ქრომატოგრაფია
HPTLC (high performance thin layer chromatography) – მაღალეფექტური თხელფენოვანი ქრომატოგრაფია
HRMS (high resolution (mass spectrometry) – მაღალი გარჩევადობა (მას-სპექტრომეტრია)
ICP-AES (inductively coupled plasma-atomic emission spectrometry) – ინდუქციურად შეწყვილებული პლაზმით ჩატარებული ატომურ-ემისიური სპექტრომეტრი
ICP-MS (inductively coupled plasma-mass spectrometry) – ინდუქციურად შეკავშირებული პლაზმური მას-სპექტრომეტრია
IR (infrared) – ინფრაწითელი
ISO (International Standard Organisation) – სტანდარტების საერთაშორისო ორგანიზაცია
LC (liquid chromatography) – სითხური ქრომატოგრაფია
LR (MS) (low resolution (mass spectrometry) – დაბალი გარჩევადობა (მას-სპექტრომეტრია)
MRPL (minimum required performance limit) – მინიმალური აუცილებელი სამუშაო ზღვარი
MS (mass spectrometry) – მას-სპექტრომეტრია
m/z (mass/charge ratio) – მასა/მუხტი, მასის თანაფარდობა მუხტთან
RF (relative migration to the solvent front (TLC)) – ფარდობითი გადაადგილება გამხსნელის ფრონტისკენ (TLC)
RSDL (relative standard deviations of the laboratory) – ლაბორატორიის ფარდობითი სტანდარტული გადახრა
SIM (selected ion monitoring) – იონების სელექტიური მონიტორინგი
TLC ( thin layer chromatography) – თხელფენოვანი ქრომატოგრაფია
UV (ultra violet light) – ულტრაიისფერი სხივი
VIS (visible light) – ხილული სხივი
5. ლიტერატურა
(1) ISO 17025: 1999 General requirement for the competence of calibration and testing laboratories. (დამკალიბრებელი და საკვლევი ლაბორატორიების კომპეტენტურობასთან დაკავშირებული ზოგადი მოთხოვნები)
(2) ISO 3534-1: 1993 Statistical Methods for quality control – Vol. 1 vocabulary and symbols. (ხარისხის კონტროლის სტატისტიკური მეთოდები – ტომი 1 ტერმინები და სიმბოლოები)
(3) ISO Guide 43-1: 1997 Proficiency testing by interlaboratory comparisons – Part 1: Development and operation of proficiency testing schemes. (ეფექტურობის კვლევა ლაბორატორიათაშორისი შედარებებით – ნაწილი 1: ეფექტურობის კვლევის სქემების შემუშავება და ფუნქციონირება.
(4) ISO Guide 43-2: 1997 Proficiency testing by interlaboratory comparisons – Part 2: Selection and use of proficiency testing schemes by laboratory accreditation bodies. (ეფექტურობის კვლევა ლაბორატორიათაშორისი შედარებებით – ნაწილი 2: ეფექტურობის კვლევის სქემების შერჩევა და გამოყენება ლაბორატორიების სააკრედიტაციო ორგანოების მიერ)
(5) ISO 5725: 1994 Accuracy (trueness and precision) of measurement methods and results – Part 1: General principles and definitions; (გაზომვის მეთოდებისა და შედეგების სისწორე (სანდოობა და სიზუსტე) – ნაწილი 1: ზოგადი პრინციპები და განმარტებები; ISO 5725-2 Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method; (ნაწილი 2: სტანდარტული გაზომვის მეთოდის განმეორებადობისა და აღწარმოებადობის განსაზღვრის ძირითადი მეთოდი; Part 4: Basic methods for the determination of the trueness of a standard measurement method. (ნაწილი 4 – სტანდარტული გაზომვის მეთოდის სანდოობის განსაზღვრის ძირითადი მეთოდი.
(6) ISO 78-2: 1999 Chemistry – Layouts for standards – Part 2: (1999 ქიმია – სტანდარტების სქემები – ნაწილი 2:) Methods of chemical analysis (ქიმიური ანალიზის მეთოდები).
(7) W.G de Ruig and J.M Weseman "A new approach to confirmation by infrared spectrometry" J. Chemometrics 4 (1990) 61-77. („ახალი მიდგომა სპექტრომეტრიით დადასტურებისადმი“)
(8) See e.g. May, T.W., Brumbaugh, W.G., 1982, Matrix modifier and L'vov platform for elimination of matrix interferences in the analysis of fish tissues for lead by graphite furnace atomic absorption spectrometry: Analytical Chemistry 54(7): 1032-1037 (90353). (იხ. მაგ. May, T.W., Brumbaugh, W.G., 1982, მატრიცის მოდიფიკატორი და L'vov-ის პლატფორმა ტყვიის შემცველობაზე თევზის ქსოვილის ანალიზისას მატრიცული დაბრკოლებების აღმოსაფხვრელად, როდესაც აღნიშნული ტარდება გრაფიტის ღუმელით ატომურ-აბსორბციული სპექტრომეტრიით: ანალიზური ქიმია 54(7): 1032-1037 (90353))
(9) Applications of Zeeman Graphite Furnace Atomic Absorption Spectrometry in the Chemical Laboratory and in Toxicology, C. Minoia, S. Caroli (Eds.), Pergamon Press (Oxford), 1992, pp. xxvi + 675. (Zeeman-ის გრაფიტის ღუმელის ატომურ-აბსორბციული სპექტრომეტრიის გამოყენება ქიმიურ ლაბორატორიაში და ტოქსიკოლოგიაში)
(10) Inductively Coupled Plasmas in Analytical Atomic Spectrometry, A. Montaser, D. W. Golighty (Eds.), VCH Publishers, Inc. (New York), 1992. (ინდუქციურად შეწყვილებული პლაზმა ანალიზურ-ატომურ სპექტრომეტრიაში)
(11) Plasma Source Mass Spectrometry Developments and Applications, G. Holland, S. D. Tanner (Eds.), The Royal Society of Chemistry, 1997, p. 329. (მიღწევები პლაზმის წყაროს მქონე მას-სპექტრომეტრიაში და მისი გამოყენება)
(12) IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies, Pure & Applied Chem, 67, 331. (მეთოდის ეფექტურობის კვლევების შემუშავების, ჩატარებისა და ინტერპრეტაციის პროტოკოლი)
(13) Jülicher, B., Gowik, P. and Uhlig, S. (1998) Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept. Analyst, 120, 173. (ნარჩენი კვალის ანალიზისას გამოყენებული გამოვლენის მეთოდების შეფასება სტატისტიკურ მონაცემებზე დაფუძნებული შიდა ვალიდაციის მეშვეობით)
(14) Gowik, P., Jülicher, B. and Uhlig, S. (1998) Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiode-array detection. Method description and comprehensive in-house validation. J. Chromatogr., 716, 221. (მრავალნარჩენიანი მეთოდი პლაზმაში არასტეროიდული ანთების საწინააღმდეგო წამლებისთვის, მაღალეფექტური სითხური ქრომატოგრაფიისა და ფოტოდიოდური მატრიცის გამოვლენის გამოყენებით. მეთოდის აღწერა და სრული შიდა ვალიდაცია)
(15) OAC-I Peer Verified Methods, Policies and Procedures, 1993, AOAC International, 2200 Wilson Blvd., Suite 400, Arlington, Virginia 22201-3301, USA. (OAC-I რეცენზირებული მეთოდები, სტრატეგიები და პროცედურები)
(16) W.J. Youden; Steiner, E.H.; "Statistical Manual of the AOAC-Association of Official Analytical Chemists", AOAC-I, Washington DC: 1975, p. 35 ff. (AOAC-ს სტატისტიკური სახელმძღვანელო)
(17) ISO 11843: 1997 Capability of detection – Part 1: Terms and definitions, Part 2: Methodology in the linear calibration case Part 2: Methodology in the linear calibration case. (გამოვლენის უნარი ნაწილი 1: ტერმინთა განმარტებები, ნაწილი 2: ხაზოვანი დაკალიბრების მეთოდოლოგია, ნაწილი 2: ხაზოვანი დაკალიბრების მეთოდოლოგია, მაგალითი.)
(18) R.W. Stephany & L.A. van Ginkel: "Yield or recovery: a world of difference". Proceedings Eight Euro Food Chem, Vienna, Austria September 18-20 (1995) Federation of European Chemical Societies, Event 206. ISBN 3-900554-17X, page 2 to 9. („მიღებული პროდუქტი თუ აღდგენა: უდიდესი სხვაობა“).
(19) 1971 წლის 18 ოქტომბრის 71/354/EEC დირექტივა საზომ ერთეულებთან დაკავშირებით წევრ სახელმწიფოებში არსებული კანონების დაახლოების შესახებ, OJ L 243, 29.10.1971, გვ. 29).
(20) ISO 31-0: 1992 რაოდენობები და ერთეულები – ნაწილი 0: ზოგადი პრინციპები
(1) მიღებული პროდუქტი: ნიმუშში არსებული საანალიზო კომპონენტის მასის ნაწილი, რომელიც გვხვდება საბოლოო ექსტრაქტში.
(2) აღდგენა (აქ): ნიმუშზე დამატებული საანალიზო კომპონენტის მასის ნაწილი, რომელიც გვხვდება საბოლოო ექსტრაქტში. დოკუმენტის დანარჩენ ნაწილში იგულისხმება, რომ მიღებული პროდუქტი და აღდგენა ერთმანეთის ტოლია და შესაბამისად გამოიყენება მხოლოდ ტერმინი „აღდგენა“.
1 ოფიციალური ბიულეტენი, L 125, 23.05.1996, გვ. 10.
2 ოფიციალური ბიულეტენი, L 65, 05.03.1998, გვ. 31.
3 ოფიციალური ბიულეტენი, L 290, 24.11.1993, გვ. 14.
4 ოფიციალური ბიულეტენი, L 286, 18.10.1990, გვ. 33.
5 ოფიციალური ბიულეტენი, L 118, 14.05.1993, გვ. 64.
6 ოფიციალური ბიულეტენი, L 118, 14.05.1993, გვ. 75.
7 ოფიციალური ბიულეტენი, L 251, 11.09.1998, გვ. 39.
დოკუმენტის კომენტარები